



















Les histiocytoses langerhansiennes résultent de la prolifération clonale d’histiocytes ayant les caractéristiques des cellules de Langerhans.
Ces histiocytes envahissent différents organes, expliquant ainsi les signes cliniques et biologiques de la maladie.
Sous le nom d’histiocytoses langerhansiennes, on regroupe désormais les maladies anciennement appelées histiocytoses X.
Histiocytes et cellules de Langerhans :
 L’histiocyte, cellule issue de la moelle osseuse, correspond à un macrophage présent à l’état quiescent dans le tissu conjonctif.
L’histiocyte, cellule issue de la moelle osseuse, correspond à un macrophage présent à l’état quiescent dans le tissu conjonctif.
Cette cellule dérive du monocyte sanguin et gagne le tissu conjonctif après la traversée de la paroi vasculaire par diapédèse.
Elle s’établit dans différents organes et sous l’effet de différentes cytokines, elle peut être activée et présenter alors des fonctions de phagocytose et de présentation antigénique.
Les histiocytes activés, suivant leur localisation tissulaire, sont les macrophages du foie (cellules de Küppfer), du poumon (macrophages alvéolaires), de l’os (ostéoclastes), etc.
La prolifération maligne de ces histiocytes est connue sous le nom de leucémie myélomonocytaire aiguë ou chronique.
En cas de prolifération avec activité phagocytaire, on parle de syndrome d’activation macrophagique.
Les cellules de Langerhans sont des cellules appartenant à la famille des cellules dendritiques qui sont localisées dans les épithélia des muqueuses buccale, anale, bronchique, du thymus et de la zone corticale des ganglions.
Ce sont des cellules qui dérivent aussi des monocytes sanguins puis migrent vers les épithélia afin de jouer le rôle de cellules « sentinelles » de l’organisme.
Au sein de l’épithélium, ces cellules vont internaliser l’antigène puis migrer vers la zone T paracorticale des ganglions lymphatiques où s’effectuera la présentation antigénique aux lymphocytes T.
Les cellules de Langerhans présentent des caractéristiques morphologiques (dendrites), phénotypiques (expression de l’antigène membranaire CD1a et de molécules HLA de classe II) et ultrastructurales (granules de Birbeck intracytoplasmiques observés au microscope électronique à transmission). L’histiocytose langerhansienne est caractérisée par la présence au sein de biopsies tumorales d’histiocytes présentant des caractéristiques de la cellule de Langerhans (antigène CD1a et granules de Birbeck).
Les anatomopathologistes parlent aussi souvent de granulomatoses à cellules de Langerhans car ces cellules siègent au sein d’un granulome d’aspect inflammatoire.
Histiocytoses langerhansiennes : fréquence, classification, causes
Les histiocytoses sont des maladies rares de l’enfant et de l’adulte jeune, touchant un peu plus souvent l’homme que la femme.
L’incidence chez l’enfant est de 1 pour 200 000, tandis que leur prévalence et leur incidence sont inconnues chez l’adulte.
On en décrit trois formes cliniques : une forme unifocale, touchant surtout l’os, appelée granulome éosinophile, une forme multifocale d’évolution chronique rassemblant des localisations osseuses crâniennes, un diabète insipide et une exophtalmie appelée maladie de Hand-Schüller-Christian et une forme multifocale de pronostic redoutable appelée maladie de Letterer-Siwe.
Des similitudes phénotypiques et ultrastructurales entre les cellules de Langerhans et les cellules tumorales provenant de ces trois formes de maladies ont conduit à abandonner le nom d’histiocytose X au profit de celui d’histiocytose langerhansienne.
Parmi les causes possibles de ces maladies, une prolifération réactionnelle en réponse à un antigène ou à une infection virale a été supposée, mais le virus ou l’antigène en cause n’ont pas été mis en évidence jusqu’à présent. Aucun argument ne permet de suspecter une maladie génétique (pas d’association connue avec un groupe HLA et très peu de cas familiaux décrits).
En revanche, nous savons depuis peu que les histiocytoses langerhansiennes sont dues à une prolifération clonale de cellules de Langerhans.
En l’absence d’anomalie du caryotype, cette prolifération clonale ne signifie pas forcément qu’il s’agit d’une prolifération maligne, mais cette mise en évidence est importante car elle justifie a posteriori l’utilisation de la chimiothérapie et de la radiothérapie dans le traitement des histiocytoses langerhansiennes.
Signes cliniques, biologiques et radiologiques :
Parmi les différentes localisations des histiocytoses langerhansiennes, les localisations osseuses sont les plus fréquentes.
Elles consistent en une atteinte des os plats et médians (os du bassin, côtes, mandibule, maxillaire, mastoïde, vertèbres) ou plus rarement des os longs, qui se manifeste par des douleurs osseuses, l’apparition d’une tuméfaction ou la survenue d’une fracture.
La radiographie montre une lacune osseuse sans aucune densification périphérique et la radiographie complète du squelette permet parfois de découvrir d’autres lésions asymptomatiques.
Satellites des localisations osseuses du crâne, des atteintes de l’audition et des complications dentaires sont fréquentes.
Des otites répétées peuvent évoluer vers une surdité et l’atteinte mandibulaire, plus fréquente que celle du maxillaire, peut provoquer chez l’enfant des troubles de la croissance des maxillaires et/ou une mobilité dentaire anormale.
Chez l’adulte, il peut exister une mobilité anormale d’une ou de plusieurs dents, une expulsion spontanée des organes dentaires ou une fracture spontanée de la mandibule.
Par ailleurs, une tuméfaction gingivale ou une ulcération muqueuse peuvent apparaître, résultant de l’extension du granulome aux tissus mous avoisinants.
Les atteintes extrasquelettiques les plus fréquentes sont un diabète insipide et une atteinte cutanéomuqueuse.
Le diabète insipide s’accompagne parfois d’un déficit d’une ou de plusieurs hormones anté-hypophysaires, qui se manifeste par une aménorrhée, une hypothyroïdie ou des troubles de la croissance générale.
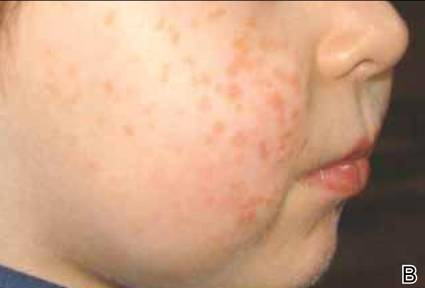
Au cours de la maladie de Letterer-Siwe, l’atteinte cutanée consiste en une éruption papuleuse de couleur jaune-orangé ou brune préférentiellement située sur le tronc, le visage et le cuir chevelu.
Une éruption maculeuse rouge orangé d’aspect séborrhéique parfois similaire à l’eczéma a aussi été rapportée.
Des ulcérations des muqueuses périnéales (anale et vulvaire) ont été rapportées chez l’adulte.
L’exophtalmie, souvent présente au cours des histiocytoses, résulte d’une infiltration rétro-orbitaire par les cellules tumorales.
Elle est le plus souvent bilatérale mais peut être asymétrique.
Cette exophtalmie peut se compliquer de cécité, par compression du nerf optique.
L’atteinte pulmonaire isolée est découverte fréquemment chez l’adulte jeune.
L’examen clinique peut retrouver également une hépatomégalie, une splénomégalie, des adénopathies, une auscultation pulmonaire anormale et une atteinte.
Il n’existe aucune anomalie biologique spécifique.
Dans les formes graves, il a été cependant rapporté une anémie et une thrombopénie lors d’atteinte médullaire et une cytolyse avec hypoalbuminémie en cas d’insuffisance hépatique.
Critères diagnostiques :
Le diagnostic est évoqué devant certains signes cliniques.
La confirmation du diagnostic nécessite une biopsie qui met en évidence des histiocytes ayant des caractéristiques spécifiques aux cellules de Langerhans, qu’elles soient morphologiques (cellules dendritiques), membranaires (expression de l’antigène CD1a) ou intracytoplasmiques (présence de granules de Birbeck).
Formes cliniques :
Le granulome éosinophile désigne une lésion osseuse lytique isolée qui peut ne pas évoluer davantage ou au contraire s’accompagner d’autres lésions osseuses, cutanéomuqueuses ou dentaires etc.
La maladie de Hand-Schüller-Christian associe une exophtalmie bilatérale, un diabète insipide et des lésions osseuses lytiques du crâne.
Elle représente une forme d’histiocytose multifocale d’évolution chronique.
Cette seule triade clinique n’est en fait présente que chez 30% des patients puisqu’il existe fréquemment des lésions supplémentaires cutanées, dentaires et de la sphère ORL associées.
La maladie de Letterer-Siwe survient le plus souvent chez le nourrisson et le petit enfant.
Cette maladie, d’évolution rapidement fatale, associe des lésions cutanées diffuses, une hépatosplénomégalie et une insuffisance médullaire, parfois associée à une insuffisance hépatique et/ou pulmonaire. L’histiocytose pulmonaire est une forme d’histiocytose décrite chez l’adulte jeune, caucasien, fumeur dans 95% des cas.
L’atteinte pulmonaire se traduit par une fatigue avec essoufflement à l’effort ou par la survenue spontanée d’un pneumothorax.
Cette maladie peut aussi être asymptomatique et découverte fortuitement devant des anomalies radiologiques telles qu’un syndrome interstitiel et des opacités nodulaires multiples de taille variable (de quelques millimètres à 1 centimètre), dont certaines apparaissent excavées.
La radiographie pulmonaire peut également montrer parfois un aspect réticulé qui témoigne de l’évolution fibrosante de la maladie, dont l’évolution extrême donne l’image dite en « rayon de miel ».
Le scanner thoracique en coupes fines est un élément important du diagnostic et montre des nodules pleins ou excavés d’aspect spiculé et la présence de kystes dont l’étendue fait toute la gravité de la maladie.
Le scanner suffit pour certains à affirmer le diagnostic dans un contexte clinique évocateur, tandis que d’autres préconisent un lavage bronchoalvéolaire.
L’évolution est imprévisible, allant de la régression spontanée et complète des lésions à la fibrose pulmonaire étendue avec insuffisance respiratoire terminale.
Aucun traitement n’a fait la preuve de son efficacité.
L’arrêt du tabac est recommandé mais il n’est pas non plus prouvé que son arrêt influence l’évolution de la maladie.
Évolution, pronostic et séquelles :
L’évolution de la maladie est le plus souvent imprévisible. Des guérisons spontanées sont possibles.
Le mauvais pronostic à court et moyen terme est lié au début précoce de la maladie et à son extension.
Les formes les plus graves débutent avant l’âge de 2 ans.
En ce qui concerne l’extension de la maladie, la gravité est davantage liée à la détérioration de la fonction d’un organe (insuffisance hépatique, insuffisance respiratoire, atteinte médullaire) qu’au nombre d’organes atteints. Plusieurs classifications sont décrites.
À long terme, le pronostic dépend essentiellement des séquelles laissées par la maladie ou par son traitement.
Les patients ont parfois une otite chronique, voire une surdité, des problèmes dentaires, des troubles de croissance ou un diabète insipide.
Les séquelles peuvent aussi être dues à la fibrose résiduelle pulmonaire (insuffisance respiratoire).
Par ailleurs, chez les patients qui survivent plus longtemps, certaines atteintes particulières peuvent survenir comme un syndrome cérébelleux invalidant ou une cholangite sclérosante.
Traitements :
Les traitements sont aussi divers que les formes cliniques de la maladie.
Actuellement, il existe des traitements focaux et des traitements généraux.
Ainsi, les lésions osseuses focales disparaissent par résection chirurgicale ou après injection locale de corticoïdes, tandis que les lésions cutanées sont traitées par l’application locale de moutardes azotées.
En cas de maladie multifocale, la chimiothérapie la plus efficace est l’étoposide.
D’autres traitements sont utilisés en milieu spécialisé : vincristine, adriamycine, cyclophosphamide, corticoïdes, etc.
Rôle du médecin généraliste :
A – Pour le diagnostic :
Sans perdre de vue la rareté de ces maladies, il faut savoir les évoquer devant certaines situations cliniques comme la survenue d’un pneumothorax spontané, une otite traînante et récidivante, des lésions cutanées atypiques.
L’histiocytose est aussi un diagnostic à suspecter devant une lacune osseuse ou un syndrome interstitiel pulmonaire inexpliqués.
B – Pour le suivi :
Les guérisons spontanées ou après traitement existent, mais il est nécessaire de rester vigilant car une récidive de la maladie est toujours possible. En cas de séquelles, le traitement au long cours le plus fréquent est le Diapidt (lypressine), hormone antidiurétique nécessaire au traitement du diabète insipide.
Dans le cas de l’histiocytose pulmonaire, une aide au sevrage tabagique est très souvent nécessaire.
Dans le cas plus difficile de maladie multifocale, les patients sont exposés aux complications du traitement par corticoïdes et de la chimiothérapie.
Enfin, en cas de traitement par le VP16t (étoposide), il existe un risque théorique de leucémie secondaire de 2 à 5% qui rend nécessaire une surveillance prolongée des patients.
Conclusion :
Les histiocytoses langerhansiennes regroupent des maladies très hétérogènes dans leurs manifestations cliniques, leur évolution et leurs traitements.
Certains signes cliniques comme la survenue d’un pneumothorax spontané ou une mobilité dentaire récente chez un patient jeune peuvent sembler banals mais doivent faire évoquer le diagnostic.
Les traitements sont aussi variés que les formes cliniques, allant de l’abstention thérapeutique à la greffe de moelle.
Les séquelles sont fréquentes en cas de forme multifocale. Une surveillance prolongée est rendue nécessaire par le risque de récidive.
En cas de guérison, l’apparition possible d’effets secondaires aux traitements et une meilleure prise en charge des séquelles de la maladie justifient un suivi prolongé.







")



{kind=link}