



















Généralités. Définitions :
Les syndromes paranéoplasiques désignent un ensemble de manifestations qui ne sont pas liées à un envahissement métastatique mais qui sont la conséquence indirecte de l’évolution d’un processus néoplasique interne.
Elles ont aussi pour particularité de suivre intimement l’histoire naturelle du cancer : apparaissant avec lui, disparaissant lors du traitement curatif du cancer, réapparaissant lors des rechutes.
Les dermatoses paranéoplasiques (DPN) semblent relativement fréquentes, puisqu’elles seraient rapportées au cours de l’évolution de 7 à 15% des cancers diagnostiqués.
 Les lésions cutanées étant directement accessibles à la vue, la reconnaissance des principaux syndromes cutanés paranéoplasiques peut permettre au clinicien d’évoquer plus rapidement la possibilité d’un cancer sous-jacent, favorisant la prise en charge d’une tumeur à un stade débutant.
Les lésions cutanées étant directement accessibles à la vue, la reconnaissance des principaux syndromes cutanés paranéoplasiques peut permettre au clinicien d’évoquer plus rapidement la possibilité d’un cancer sous-jacent, favorisant la prise en charge d’une tumeur à un stade débutant.
Par ailleurs, les lésions cutanées observées au cours des DPN sont parfois très affichantes, voire gênantes et le contrôle de la maladie cancéreuse peut permettre d’améliorer la qualité de la vie des patients.
Plusieurs critères caractérisent une DPN.
– Des critères cliniques.
La DPN est une entité clinicopathologique spécifique et précise, facilement identifiable et orientant d’emblée vers un cancer associé donné.
– Des critères chronologiques.
La DPN doit survenir uniquement après le développement de la tumeur.
La DPN peut être diagnostiquée avant que le cancer n’ait été dépisté, et c’est l’intérêt de la connaître pour le diagnostic précoce du cancer, mais le développement du cancer précède la DPN. L’évolution de la DPN accompagne étroitement celle favorable ou défavorable du cancer.
Autrement dit encore, la dermatose et la tumeur ont le même profil évolutif : l’ablation ou tout autre moyen de mise en rémission du cancer aboutit à la régression de la dermatose, alors que la récidive du cancer ou la survenue de métastases s’associent à une reprise évolutive des manifestations cutanées.
– Des critères étiopathogéniques.
La DPN ne connaît habituellement pas d’autre étiologie que le cancer.
Dans l’idéal, chaque DPN devrait répondre à un mécanisme pathogénique clair et identifié et devrait avoir une prédilection pour un type histologique particulier de cancer.
Autrement dit, on pourrait supposer une relation directe de cause à effet entre la DPN et le cancer profond.
Classifications :
Plusieurs classifications des DPN peuvent être proposées : classification selon les lésions élémentaires qui est celle que nous adopterons pour décrire les principaux tableaux, classification selon le mécanisme pathogénique, selon la chronologie d’apparition de la DPN par rapport au diagnostic du cancer, etc.
Cependant, aucune n’est réellement satisfaisante car les DPN se caractérisent avant tout par leur grande hétérogénéité, ce qui rend impossible toute tentative de classification cohérente et logique.
On a même une certaine impression d’amalgame ou de confusion dans la littérature.
Enfin, on distingue aussi une classification de type « probabiliste » qui est celle qui tient le mieux compte de la spécificité et de la fréquence de l’association entre la DPN et le cancer et qui isole des DPN vraies, des DPN facultatives et des DPN dont la réalité est plus controversée car le plus souvent fortuites.
A – SYNDROMES PARANÉOPLASIQUES CUTANÉS VRAIS :
Il s’agit de phénomènes cutanés spécifiquement associés à un cancer et qui suivent intimement son évolution.
Le tableau clinique est stéréotypé et va permettre de poser d’emblée à la fois un diagnostic de DPN et d’évoquer la présence d’un cancer profond.
Autrement dit, une telle dermatose est tellement spécifique qu’elle n’a pas d’autre étiologie que le cancer.
Le syndrome paranéoplasique est ainsi un véritable marqueur du cancer qui doit être recherché avec acharnement.
Et c’est dans ces circonstances qu’il faut pratiquer un bilan exhaustif.
Seul un petit nombre de dermatoses répondent stricto sensu à cette définition.
Bien qu’elles soient rares, leur association quasi obligatoire à un cancer spécifique suggère que ce rapprochement n’est pas le fruit du hasard mais résulte bien d’une relation directe entre ces deux processus pathologiques.
Cependant, même dans ce cadre restrictif, certaines DPN peuvent être rapportées au cours de l’évolution de différents types de cancers : par exemple le pemphigus paranéoplasique a été décrit en association avec des lymphomes ou des hémopathies lymphoïdes chroniques mais aussi au cours de l’évolution de sarcomes ou de cancers pulmonaires.
B – SYNDROMES PARANÉOPLASIQUES NON SPÉCIFIQUES OU FACULTATIFS :
Un grand nombre de manifestations cutanées sont occasionnellement observées au cours de certains cancers sans répondre stricto sensu aux critères qui définissent une DPN.
Il s’agit de dermatoses fréquentes et non spécifiques qui ont parfois été décrites en association avec certaines néoplasies profondes mais qui apparaissent habituellement de manière isolée, ou qui coexistent avec des pathologies bénignes.
C’est le cas par exemple des vascularites qui relèvent habituellement d’une cause infectieuse, médicamenteuse ou dysimmunitaire, de l’ichtyose acquise qui peut être aussi d’origine médicamenteuse ou carentielle, du syndrome des ongles jaunes qui associe une xanthonychie, des oedèmes des membres inférieurs et des épanchements pleuraux et qui serait consécutif à des processus intrathoraciques qui sont habituellement bénins (bronchectasies, anomalies du réseau lymphatique, etc).
De la même manière, le caractère paranéoplasique des érythèmes annulaires centrifuges est probablement exceptionnel. Ces DPN facultatives semblent moins spécifiques d’un type donné de cancer.
Ainsi, par exemple, l’ichtyose acquise a été décrite au cours des hémopathies, des cancers du sein, du poumon, des cancers digestifs, etc.
Par ailleurs, plusieurs DPN différentes peuvent apparaître de manière conjointe ou successive au cours de l’évolution d’un seul cancer.
C – ASSOCIATIONS DERMATOSES-CANCER :
La pertinence de certaines associations dermatose-cancer est discutable car la survenue conjointe d’une dermatose et d’une tumeur peut être tout à fait fortuite.
Ce cadre représente quantitativement probablement la majorité des lésions pour lesquelles le caractère « paranéoplasique » est attribué par excès, et qui s’explique par la grande fréquence de certaines dermatoses.
Ainsi, les relations sclérodermies-cancer sont douteuses, et la fréquence des cancers au cours de cette collagénose est tout à fait identique à celle de l’ensemble de la population.
Dans d’autres cas, la dermatose peut avoir simplement aussi simulé une évolution selon un mode paranéoplasique par sa chronologie de survenue par rapport à la tumeur.
Dans d’autres cas aussi, la dermatose et le cancer partagent une étiologie commune : ainsi, la porphyrie cutanée tardive (PCT) est souvent associée à une infection par le virus de l’hépatite C, et cette infection se complique fréquemment de cirrhose et d’hépatocarcinome, mais la PCT n’est pas un syndrome paranéoplasique révélateur du cancer du foie.
Ainsi aussi, la dermatite herpétiforme est associée à une entéropathie au gluten qui se complique souvent de lymphome digestif, mais la dermatite herpétiforme n’est pas stricto sensu un syndrome paranéoplasique.
Seuls les outils épidémiologiques comme les études de type castémoin peuvent vérifier la validité de certaines de ces associations.
Les études cas-témoin remettent ainsi en question l’existence du signe de Leser-Trélat.
D – ANOMALIES CUTANÉES EN RELATION AVEC DES CANCERS PROFONDS MAIS NON CONSIDÉRÉES COMME DES DERMATOSES PARANÉOPLASIQUES :
Un grand nombre de lésions cutanées peuvent être décrites lors de l’évolution d’un cancer et elles relèvent de mécanismes divers non paranéoplasiques.
– Lésions qui sont la conséquence directe de l’évolution du processus néoplasique, résultant par exemple d’un phénomène d’envahissement métastatique à la peau ou d’une extension par contiguïté à la peau du processus néoplasique, etc.
– Lésions secondaires à divers dépôts dans la peau (ictère, mélanose, amyloïdose, etc) ou à différentes sécrétions ou produits de la tumeur ayant un impact sur la peau (flush, prurit, etc), encore que stricto sensu certaines de ces lésions répondent à certains des critères caractérisant une DPN.
– Lésions cutanées secondaires à une complication lors de l’évolution du cancer : phénomènes compressifs ou complications des traitements du cancer : radiodermite, herpès, infections bactériennes, etc.
– Dermatoses non spécifiques secondaires à des anomalies systémiques variées causées par le cancer : malnutrition, etc.
D’autres lésions cutanées s’intègrent dans le cadre des génodermatoses.
Certaines lésions cutanées sont intégrées dans des syndromes habituellement héréditaires et bien individualisés (génodermatoses) au cours de l’évolution desquels surviennent de façon quasi obligatoire un ou plusieurs cancers profonds.
Les lésions cutanées sont alors un marqueur de la prédisposition des individus atteints au développement de néoplasie.
Elles précèdent les tumeurs internes mais elles ne sont pas directement le témoin de l’apparition du cancer, ou ne sont pas causées par lui.
Au sein d’une même famille, l’ensemble des individus atteints de ces lésions cutanées doit bénéficier d’un suivi régulier et orienté afin de dépister rapidement l’apparition d’un cancer au cours de la vie.
À l’inverse, par définition, les sujets atteints de DPN ne sont pas génétiquement prédisposés à développer des cancers et il n’y a pas de risque familial.
Mécanismes des dermatoses paranéoplasiques :
Bien que ce soit la tumeur qui soit à l’origine des lésions cutanées, les mécanismes sont probablement multiples et intriqués, ce qui expliquerait les associations privilégiées qui semblent exister entre certaines DPN pour des cancers donnés.
Globalement, ces mécanismes ne sont toujours pas bien appréhendés.
À l’échelon tissulaire, le fait qu’une même DPN peut être retrouvée dans des cancers histologiquement très différents suggère que des lignées cellulaires d’origine variée pourraient se dédifférencier ou se redifférencier en un même état fonctionnel intermédiaire devenant métaboliquement actif.
À l’échelon cellulaire, les lésions seraient expliquées par l’activation d’oncogènes ou la perte d’expression des protéines inhibant les oncogènes, favorisant l’activation ou la production de différentes cytokines ou de certaines substances biologiquement actives agissant à distance sur des organes-cibles.
On peut évoquer les différents mécanismes pathogéniques possibles indiqués ci-après.
A – PROCESSUS CARENTIEL :
– Déplétion, détournement ou surconsommation de différentes substances par la tumeur entraînant un tableau de carence ayant un retentissement cutané par troubles de la synthèse des protéines de l’épiderme : surconsommation de tryptophane et perte accrue par les diarrhées, responsables des dermatoses pellagroïdes des tumeurs carcinoïdes, surconsommation en tryptophane qui intervient aussi dans la synthèse du glucagon au cours de l’érythème nécrolytique migrateur des cancers du pancréas, etc.
B – PROCESSUS HORMONAL OU HUMORAL :
– Production par la tumeur de substances hormonales ou à action hormonale, directement responsable des manifestations cutanées mais agissant à distance : sécrétion et libération de peptides vasoactifs comme la sérotonine, la kallicréine, l’histamine par les tumeurs carcinoïdes, sécrétion d’hormone antidiurétique (ADH) par les cancers à petites cellules pulmonaires, libération ectopique par la tumeur (carcinome bronchique) de peptides à action adrenocorticotrophic hormone (ACTH)-like et b-melanocyte stimulating hormone (b-MSH)-like au cours des syndromes de Cushing paranéoplasiques, production exagérée et ectopique de growth hormone (GH), thyroid stimulating hormone (TSH), MSH, ACTH par la tumeur au cours des acanthosis nigricans malins, production exagérée de GH au cours de certaines kératodermies palmoplantaires.
Mais l’identification de ces polypeptides reste exceptionnelle.
– Sécrétion accrue et libération systémique de diverses enzymes pancréatiques (amylase, lipase, etc) qui sont responsables des panniculites nodulaires avec adiponécrose (syndrome de Weber-Christian) observées au cours des carcinomes du pancréas, etc.
C – LIBÉRATION DE CYTOKINES ET DE FACTEURS DE CROISSANCE :
– Sécrétion par la tumeur de facteurs de croissance ou de facteurs équivalents dans les DPN prolifératives : epidermal growth factor (EGF), transforming growth factor (TGF), insulin-like growth factor 1 (IGF1) au cours des acanthosis nigricans, de certaines ichtyoses, de certaines kératodermies palmoplantaires, de l’hippocratisme digital et de l’ostéoarthropathie pneumique hypertrophiante, de l’éruption de kératoses séborrhéiques (KS) profuses (signe de Leser- Trélat), etc.
D – DIVERS PROCESSUS IMMUNOLOGIQUES :
– Production par la tumeur de superantigènes bactériens capables de se fixer avec les récepteurs du complexe majeur d’histocompatibilité induisant l’activation et la prolifération de lymphocytes T spécifiques.
– Production par la tumeur d’haptènes tumoraux qui ont une antigénicité croisée avec divers composants de la peau induisant une rupture de tolérance et la fixation de ces anticorps sur des antigènes épithéliaux dans le pemphigus paranéoplasique.
– Immunité lymphocytaire de type cytotoxique dirigée contre les antigènes tumoraux, mais aussi contre des composants musculaires et cutanés au cours de la dermatomyosite.
– Rupture du processus de « non-contact » entre les cellules épithéliales, mésenchymateuses et neuroendocrines, par défaillance du dispositif mésenchymateux immunologique favorisant les échanges moléculaires normalement réprimés et l’activation des lymphocytes.
Cas particulier des dermatoses au cours des hémopathies :
Les hémopathies s’accompagnent très fréquemment de manifestations cutanées.
Les lésions sont soit en relation avec la cytopénie (pâleur, purpura, etc), soit liées à la localisation et à la prolifération au niveau de la peau des cellules hématopoïétiques malignes, soit liées à l’immunodépression assez spécifique de ces contextes, soit liées à un syndrome paranéoplasique.
Les DPN au cours des hémopathies ont des mécanismes physiopathogéniques relativement spécifiques : dépôts d’immunoglobulines (Ig) ou de complexes immuns circulants sécrétés par la tumeur, activation des polynucléaires par différentes cytokines circulantes (granulocyte-colony stimulating factor [G-CSF]), autres effets immunologiques de ces Ig, syndrome d’hyperviscosité sanguine par agrégation et activation plaquettaire responsable d’une obstruction artériolaire au cours de l’érythermalgie des syndromes myéloprolifératifs, etc.
Mais la majeure partie des DPN décrites au cours des hémopathies ne sont pas spécifiques puisqu’elles ont aussi été décrites avec des tumeurs solides, ou particulièrement pour les dermatoses neutrophiliques au cours de l’évolution de maladies inflammatoires, rhumatismales ou digestives.
On peut malgré tout individualiser quelques liens étroits et relativement spécifiques.
C’est le cas par exemple des érythrodermies et des dermatoses exfoliatrices survenant au cours de l’évolution des lymphomes cutanés T, des myélodysplasies ou des leucémies, ou celui des vascularites cutanées au cours des syndromes myélodysplasiques (leucémie aiguë à tricholeucocytes).
C’est le cas aussi des dermatoses neutrophiliques : syndrome de Sweet qui peut annoncer l’acutisation du syndrome myélodysplasique, pyoderma gangrenosum au cours du myélome ou des leucémies aiguës.
L’aspect chronologique est assez particulier : les manifestations cutanées surviennent habituellement au cours de l’évolution de l’hémopathie, quand celle-ci a déjà été diagnostiquée.
Principales dermatoses paranéoplasiques :
A – DÉSORDRES MUSCULOSQUELETTIQUES :
1- Syndrome de pachydermopériostose :
Il correspond à une hypertrophie des différents composants de la peau (épiderme épaissi, collagène hypertrophié), de ses annexes (hyperséborrhée et hyperhidrose) et des extrémités osseuses.
La pachydermopériostose peut être primitive autosomique dominante (expressivité variable) ou secondaire paranéoplasique.
Le tableau clinique est habituellement localisé, réalisant au niveau des mains un hippocratisme digital, au niveau des extrémités des membres un tableau d’ostéoarthropathie hypertrophiante, au scalp celui de cutis verticis gyrata ou pachydermie plicaturée.
Si la réalité du caractère paranéoplasique de l’hippocratisme digital et de l’ostéoarthropathie hypertrophiante (qui coexistent dans 10 à 20 % des cas et qui seraient deux stades différents d’une même affection) est indiscutable, la majorité des observations de cutis verticis gyrata seraient génétiquement déterminées ou d’origine postinflammatoire alors que les cas paranéoplasiques seraient exceptionnels.
Mécanisme : les manifestations seraient liées à la sécrétion de facteurs de croissance, dont le transforming growth factor (TGF) a.
* Hippocratisme digital :
Clinique : il se caractérise par une hypertrophie symétrique des phalanges distales associée à une déformation des ongles bombés en « verre de montre ».
Une cyanose peut venir compléter le tableau.
Cancers associés : dans le contexte de DPN, les manifestations prédominent chez l’homme d’âge moyen, et sont associées à un carcinome bronchopulmonaire, plus rarement à un mésothéliome, un carcinome de l’estomac, de l’oesophage.
Souvent, les lésions apparaissent lorsque le cancer est à un stade déjà avancé.
Mécanisme : l’épaississement résulte d’une augmentation du flux sanguin périphérique (trouble primitif de la circulation pulmonaire ?) avec oedème périunguéal, responsable d’anomalies de l’oxygénation tissulaire locale, d’une dilatation des vaisseaux des extrémités et d’une stase des éléments figurés.
Particularités : l’hippocratisme digital peut aussi être héréditaire et il a été décrit au cours de nombreuses affections bénignes cardiorespiratoires (abcès, bronchopneumopathies obstructives chroniques, bronchectasies…) et au cours des cirrhoses.
* Ostéoarthropathie hypertrophiante :
Clinique : l’ostéoarthropathie hypertrophiante associe des extrémités acromégaloïdes ou « mains en battoir » à des manifestations articulaires et osseuses douloureuses des os longs et des grosses articulations.
Les manifestations osseuses pseudo-inflammatoires et très douloureuses correspondent à une périostite proliférante bilatérale avec ossification périostée irrégulière dont les signes radiologiques sont pathognomoniques : présence d’une fine ligne transparente entre une corticale osseuse épaissie et une réaction périostée.
Cancers associés : l’ostéoarthropathie hypertrophiante est pratiquement toujours associée à un carcinome bronchique, plus rarement à d’autres tumeurs intrathoraciques ou à des métastases pulmonaires de tumeurs d’origine variée. Particularités : une gynécomastie est signalée dans 10 % des cas.
Les douleurs osseuses ne répondent pas à la colchicine ou aux antiinflammatoires non stéroïdiens (AINS) mais plutôt aux diphosphonates, à l’octréotide ou à la radiothérapie.
2- Dermatomyosite :
Clinique : un rash héliotrope, une myopathie proximale et des papules sur les phalanges proximales sont caractéristiques du tableau.
La survenue après 60 ans, chez un homme, un caractère érosif nécrotique ou ulcéré des lésions cutanées, l’existence d’une fibrose pulmonaire, la présence de nécroses musculaires ou d’une vascularite leucocytoclasique à l’histologie, un syndrome inflammatoire biologique majeur (vitesse de sédimentation > 40 à la première heure) et des taux élevés de créatine phosphokinase sérique doivent faire évoquer le caractère paranéoplasique de la dermatomyosite.
Cancers associés : la dermatomyosite serait associée à un cancer dans 20 à 30 % des cas.
Chez l’homme, les localisations tumorales sont par ordre de fréquence les cancers bronchiques et digestifs (estomac et côlon), chez la femme les cancers gynécologiques prédominent (tumeurs ovariennes).
En Asie et en Tunisie, la localisation préférentielle du cancer au niveau de la sphère ORL, en particulier le cavum, est à mentionner. Particularités : les cancers sont habituellement diagnostiqués de façon concomitante à la dermatomyosite ou dans les mois qui suivent ou précèdent le tableau cutané.
La dermatomyosite amyopathique, sans atteinte musculaire inflammatoire, peut aussi s’associer avec un cancer.
Le traitement des manifestations cutanées et musculaires fait appel aux corticoïdes et aux immunosuppresseurs et il n’aurait pas d’influence sur le développement de la néoplasie et sur son cours évolutif.
Le traitement de la néoplasie sous-jacente améliore aussi la dermatomyosite.
3- Fasciite palmaire et polyarthralgies :
Clinique : l’âge de survenue est généralement supérieur à 55 ans, avec une nette prédominance féminine.
Le tableau touche de façon symétrique les mains et les doigts, plus rarement les plantes.
On observe des arthralgies avec raideur matinale, un oedème digital et palmaire limitant les amplitudes et évoluant rapidement vers une induration, une rétractation irréductible des doigts.
Une cyanose sans syndrome de Raynaud, une hyperhidrose ainsi que des arthralgies des épaules peuvent compléter le tableau.
Les radiographies montrent une simple déminéralisation.
Le bilan sanguin immunologique (latex, Waaler-Rose, ACAN) est le plus souvent normal et le syndrome inflammatoire modéré.
Histologie : elle montre une fibrose au niveau du derme et du fascia associée à un infiltrat mononucléé périvasculaire habituellement modéré.
Cancers associés : la nature paranéoplasique de cette entité est incertaine.
Dans les cas publiés de néoplasies associées, 50 % étaient des adénocarcinomes ovariens.
On trouve également des cancers du pancréas, côlon, vessie, poumon, etc.
La fasciite palmaire survient souvent au stade métastatique, d’où un pronostic péjoratif.
Mécanismes : la présence d’IgG en immunofluorescence directe (IFD) inconstamment retrouvée au niveau de l’aponévrose palmaire est en faveur d’une origine auto-immune.
Certains auteurs supposent l’existence d’une substance circulante sécrétée par la tumeur et induisant une prolifération fibroblastique.
Enfin, les similitudes avec l’algodystrophie font discuter une anomalie du système sympathique.
B – ÉRYTHÈMES RÉACTIFS :
1- Érythème nécrolytique migrateur :
Clinique : le tableau se caractérise par des placards érythémateux et circinés, limités par une collerette desquamative, qui évoluent de façon centrifuge.
Les lésions évoluent par poussées successives et peuvent devenir vésiculobulleuses puis croûteuses. Elles régressent spontanément en quelques semaines en laissant des placards hyperpigmentés.
La topographie est surtout périorificielle : périnée, face ; et distale : extrémités des membres. Une stomatite, une glossite douloureuse et une perlèche peuvent accompagner le tableau cutané.
Une altération de l’état général avec amaigrissement et l’apparition brutale d’un diabète insulinodépendant sont décrites dans plus de 90 % des cas, plus rarement une anémie, une démence ou des manifestations digestives avec diarrhée, douleurs.
Le risque thromboembolique est considéré comme élevé. Le bilan biologique confirme une intolérance glucidique, avec un taux plasmatique élevé de glucagon.
Le test à l’arginine (doublement rapide du taux de glucagon après administration d’arginine) est positif. Histologie : on observe une parakératose surmontant une dégénérescence vacuolaire du tiers supérieur de l’épiderme avec oedème et nécroses kératinocytaires alors que la partie inférieure de l’épiderme est conservée (réalisant l’image de « tranche napolitaine »).
Il existe un infiltrat polymorphe périvasculaire du derme et l’IFD est négative.
Cancers associés : l’érythème nécrolytique migrateur est le plus souvent lié à un glucagonome qui est développé aux dépens des îlots a de Langerhans et localisé dans la moitié des cas au niveau de la queue du pancréas.
Le glucagonome peut être sporadique ou s’intégrer dans un contexte de néoplasies endocriniennes multiples (NEM).
Dans 50 % des cas, il existe des métastases au moment du diagnostic (hépatiques ou ganglionnaires), et la survie médiane au moment du diagnostic est de 2 ans.
Particularités : la fréquence de l’association entre érythème nécrolytique et glucagonome est proche de 100 %, mais des cas sans glucagonome ont été rapportés (24 cas décrits en 1998).
Il s’agit le plus souvent de maladie coeliaque, ou d’autres syndromes avec malabsorption ou d’hépatopathies chroniques (cirrhose ou hépatites), mais aussi d’autres tumeurs.
Le traitement est chirurgical plus ou moins associé à une chimiothérapie (streptozocine ou dacarbazine).
Le traitement par somatostatine est symptomatique et transitoire.
Mécanisme : le rôle précis du glucagon dans la dermatose n’est pas établi mais semble passer par un hypercatabolisme entraînant une hypoaminoacidémie.
En effet, on note une amélioration des signes cutanés après perfusion d’aminoacides ainsi qu’une remontée des taux d’aminoacides après traitement du glucagonome.
Le rôle d’un déficit en zinc reste à établir.
2- Erythema gyratum repens :
Clinique : l’éruption est constituée de bandes érythémateuses de 1 à 3 cm de large, concentriques, serpigineuses, réalisant un aspect de vagues ou de « nervures de bois » rapidement migratrices (jusqu’à 1 cm/j).
Ces bandes présentent une collerette desquamative et sont habituellement très prurigineuses.
Les lésions siègent préférentiellement sur le tronc et la racine des membres et épargnent habituellement la face et les extrémités des membres.
Le sex-ratio serait de deux hommes pour une femme et l’âge moyen de survenue est de 63 ans. Une hyperéosinophilie est inconstamment notée.
Histologie : elle est non spécifique ; hyperkératose légère à modérée, parfois spongiose et parakératose focale, associées à un infiltrat lymphohistiocytaire périvasculaire sans vascularite.
En IFD, des dépôts granulaires d’IgG et de complément ont été parfois constatés au niveau de la membrane basale.
Cancers associés : sur 49 cas recensés par Boyd en 1992, seuls sept cas n’étaient pas associés à un cancer sous-jacent, soit une fréquence de l’association de 85 %.
Le plus souvent, l’éruption précède le diagnostic de cancer (7 mois en moyenne) et l’évolution cutanée est parallèle à la néoplasie associée.
Les cancers les plus fréquemment associés sont les carcinomes pulmonaires (32 %), les carcinomes oesophagiens ou mammaires sont plus rares (6 à 7 %).
Il s’associe parfois à cette éruption serpigineuse une hyperkératose palmoplantaire ou une ichtyose.
Des observations ponctuelles en association avec un syndrome CREST, une tuberculose pulmonaire ont été rapportées.
Mécanisme : la découverte par plusieurs auteurs de dépôts d’Ig et de complément au niveau de la membrane basale en peau lésée fait évoquer une hypothèse immunologique : réaction croisée entre des antigènes constitutifs de la membrane basale bronchique altérée et des antigènes de la membrane basale cutanée.
3- Syndrome carcinoïde :
Clinique : les manifestations paroxystiques, souvent déclenchées par les aliments épicés, l’alcool, l’exercice, certains médicaments, associent bouffées vasomotrices (flush dans la région céphalique), sueurs, larmoiements, érythème facial.
À long terme, elles peuvent aboutir à des modifications sclérodermiformes de la peau, des télangiectasies et un érythème violacé du visage de type pellagroïde.
Les manifestations extradermatologiques peuvent compléter cette sémiologie : tachycardie, malaise, diarrhée motrice, toux ou dyspnée asthmatiforme, insuffisance cardiaque droite progressive par fibrose de l’endocarde, etc.
Le diagnostic se confirme par le dosage de la sérotonine circulante et de divers de ses métabolites urinaires tels que l’acide 5-hydroxy-indol-acétique.
Cancers associés : dans le contexte de tumeur maligne, les manifestations paroxystiques évoluent sur un mode aigu mais le diagnostic est souvent porté à un stade tardif lorsque la tumeur, qui est habituellement localisée au niveau de l’intestin grêle, du rectum, du poumon, des ovaires, du pancréas s’est déjà compliquée de localisations secondaires habituellement hépatiques.
Mécanisme : il résulte de la sécrétion en grande quantité et du passage dans la circulation générale de peptides vasoactifs type sérotonine, prostaglandines, vaso-intestinal peptide (VIP), bradykinines et kallicréines à partir d’une tumeur neuroendocrine développée aux dépens du système amine precursor uptake decarboxylation (APUD).
Les autres étiologies classiques des flushes sont à éliminer (médicaments…).
L’octréotide pourrait améliorer les manifestations paroxystiques.
4- Érythrodermies :
Les érythrodermies ont été majoritairement décrites au cours des hémopathies : lymphomes cutanés ou systémiques, plus rarement myélodysplasies ou leucémies.
D’autres cancers (adénocarcinomes, carcinomes épidermoïdes, mélanome) ont été décrits en association, mais la relation de cause à effet n’est pas toujours bien documentée.
C – DÉSORDRES VASCULAIRES :
Thrombophlébites migrantes ou phlébites de Trousseau :
Clinique : des localisations inhabituelles (membre supérieur), des épisodes de thromboses à la fois artérielles et veineuses, l’importance de la réaction inflammatoire associée et une évolution prolongée précédant de plusieurs années le diagnostic de cancer caractérisent cette affection.
Cancers associés : les phlébites migratrices superficielles et récurrentes sont en relation avec des cancers gastriques, pancréatiques, pulmonaires ou génitaux.
Mécanisme : il reste mal connu ; activation plaquettaire, production de facteurs activant l’hémostase, rôle des anticorps antiphospholipides, etc.
D – DERMATOSES SQUAMEUSES ET PROLIFÉRANTES :
Toutes sont caractérisées par un épaississement de l’épiderme : soit par hyperkératose (acanthosis nigricans, ichtyose, pityriasis rotunda, pachydermatoglyphie), soit par acanthose (syndrome de Bazex), soit par l’association des deux processus (papillomatose floride, signe de Leser-Trélat).
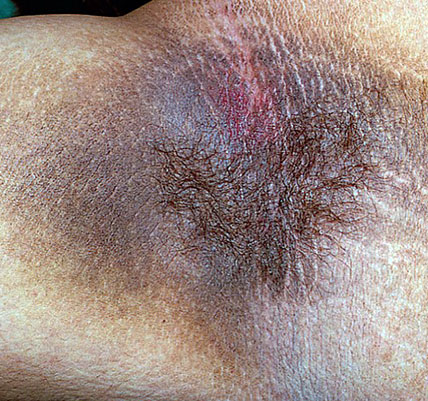
1- Acanthosis nigricans :
Clinique : il est caractérisé par un épaississement de la peau et se présente sous la forme de lésions brunâtres parfois papillomateuses, d’aspect sale et qui se localisent de façon symétrique au niveau des plis axillaires, de la nuque et des faces latérales du cou, mais aussi du périnée, des régions antécubitales et poplitées ainsi que des aréoles mammaires.
On distingue classiquement l’acanthosis nigricans malin (paranéoplasique) de l’acanthosis nigricans bénin, qui peut être familial, endocrinien, médicamenteux (corticoïdes, acide nicotinique, acide fusidique, oestrogènes, insuline) ou idiopathique.
La survenue rapide, extensive, chez un sujet d’âge mûr, les localisations muqueuses (langue, lèvres, paupières) et périorbitaires, voire le caractère prurigineux, sont évocateurs d’une origine paranéoplasique.
D’autres dermatoses, que certains auteurs veulent intégrer dans un unique continuum, peuvent s’associer à l’acanthosis malin : la papillomatose cutanée floride, les anomalies palmoplantaires (kératodermie et pachydermatoglyphie), le signe de Leser-Trélat.
Histologie : elle ne permet pas de différencier l’acanthosis nigricans malin de l’acanthosis nigricans bénin.
Elle montre une hyperkératose orthokératosique, une papillomatose, des zones d’acanthose alternant avec des zones d’atrophie épidermique, une augmentation des pigments mélaniques dans le stratum corneum.
Cancers associés : la néoplasie sous-jacente est principalement abdominale (90 %) et est représentée plus spécialement par un adénocarcinome gastrique (64 %).
Les autres cancers associés sont utérins, hépatiques, colorectaux, ovariens, etc.
Concrètement, l’évolution de l’acanthosis nigricans est corrélée à celle de la néoplasie associée qui est déjà souvent à un stade avancé. Physiopathologie : l’expression de facteurs de croissance par la tumeur pourrait être incriminée.
En effet, plusieurs auteurs ont retrouvé des expressions cutanée et tumorale de récepteurs pour l’EGF, impliqués dans la prolifération et la différenciation des divers épithéliums, ainsi que des taux élevés de TGF a tantôt sanguins, tantôt urinaires.
2- Ichtyose :
Clinique : il s’agit d’une affection à prédominance masculine, cliniquement proche de l’ichtyose vulgaire mais qui s’en différencie par un âge d’apparition tardif, l’absence d’antécédents familiaux, le caractère inflammatoire (« aspect d’ichtyose rouge »), l’extension aux plis de flexion, l’atteinte palmoplantaire et le prurit souvent associés. Histologie : elle est proche de celle décrite dans l’ichtyose vulgaire : hyperkératose orthokératosique modérée, amincissement de la granuleuse, discret infiltrat périvasculaire.
L’étude ultrastructurale met en évidence une réduction de la synthèse de kératohyaline dont la structure reste normale, contrairement à l’ichtyose vulgaire dominante.
Cancers associés : la maladie de Hodgkin représenterait 70 à 80 % des cas d’ichtyose paranéoplasique.
L’ichtyose est habituellement concomitante ou postérieure au diagnostic de la maladie de Hodgkin mais elle peut précéder le diagnostic de 1 an ou plus.
L’ichtyose associée aux lymphomes non hodgkiniens est plus rare et les lésions cutanées sont contemporaines de la rechute du lymphome.
Elle a également été décrite en association avec d’autres hémopathies : myélome multiple, leucémies, mycosis fongoïde et plus rarement avec des carcinomes du poumon, côlon, sein, des sarcomes de Kaposi.
Particularités : elle peut coexister avec d’autres DPN comme l’acrokératose de Bazex, l’acanthosis nigricans.
Par ailleurs, des tableaux similaires ont été décrits au cours de maladies variées qu’il conviendra d’éliminer avant de porter le diagnostic de DPN : causes médicamenteuses (hypocholestérolémiants), endocriniennes (hypothyroïdie), carences en vitamine A, états de dénutrition, infections à virus d’immunodéficience humaine (VIH).
Mécanismes : la cellule tumorale produirait différents facteurs de croissance ; le rôle du TGF a qui module le métabolisme lipidique est particulièrement discuté.
3- Pityriasis rotunda :
Clinique : cette affection est habituellement décrite dans les populations asiatiques et noires sud-américaines alors qu’elle est rare en Europe.
L’homme d’âge moyen est le plus souvent concerné.
La lésion élémentaire est finement squameuse, asymptomatique, réalisant des plaques circulaires non inflammatoires bien limitées, à bords nets, dont la teinte est uniforme et parfois pigmentée.
Le nombre et la taille des lésions sont variables, elles peuvent confluer sur le tronc, les épaules, les cuisses donnant un aspect géométrique évocateur. Histologie : hyperkératose orthokératosique, avec un amincissement de la couche granuleuse.
Cancers associés : le pityriasis rotunda prédominerait en association avec des adénocarcinomes hépatiques dans 6 % des cas et avec des adénocarcinomes gastriques, mais la majorité des lésions évoluent sans cancer associé : tuberculose, cirrhose ou dans des contextes de dénutrition.
Il existe aussi des formes familiales.
Mécanismes : la présence d’une hyperkératose et d’une atrophie de la couche granuleuse suggère qu’un grand nombre de ces lésions s’apparentent probablement à une forme localisée d’ichtyose.
4- Pachydermatoglyphie ou « tripe palms » :
Clinique : dans une revue de la littérature de 1993, PR Cohen fait la synthèse des 87 cas publiés de cette entité qu’on rapporte indifféremment sous plusieurs noms : acanthosis palmaris, pachydermatoglyphie ou « tripe palms » dans le vocable anglosaxon en référence à l’aspect de la muqueuse bovine.
Ces termes traduisent indifféremment un épaississement rugueux des paumes et parfois des plantes, avec un aspect velouté et une coloration jaunâtre.
Cette apparence villeuse correspond à une accentuation des dermatoglyphes de la face palmaire des mains.
La pachydermatoglyphie serait associée dans 75 % des cas à un acanthosis nigricans.
Certains auteurs pensent d’ailleurs que la pachydermatoglyphie n’est qu’une manifestation palmaire de cette dermatose. Histologie : bien que rarement réalisée, elle montre une hyperkératose et une acanthose.
Plus rarement, ont été rapportés une papillomatose, une mucinose dermique et une augmentation des mastocytes dermiques.
Cancers associés : une néoplasie est retrouvée dans 90 % des cas.
Les signes cutanés précèdent ou accompagnent la néoplasie dans deux tiers des cas.
Si la pachydermatoglyphie est associée à un acanthosis nigricans, le cancer le plus souvent retrouvé est le cancer gastrique, avec une fréquence supérieure à celle retrouvée en cas de pachydermatoglyphie isolée (35 % contre 12 %).
En revanche, en cas de pachydermatoglyphie isolée, le cancer le plus souvent associé est pulmonaire (53 %).
Viennent ensuite les cancers gastriques (12 %), vésicaux, mammaires, rénaux, ovariens…
Mécanismes : le rôle de l’hormone de croissance (GH) a été initialement décrit mais les hypothèses actuelles évoquent la sécrétion par la tumeur de facteurs de croissance kératinocytaires stimulant l’hyperplasie épidermique : des taux élevés de TGF a, d’EGF ont parfois été mesurés.
5- Hyperkératose palmaire filiforme acquise :
Clinique : multiples lésions acuminées filiformes kératosiques et spiculées localisées à la face palmaire des deux mains et des doigts.
Histologie : les anomalies sont essentiellement épidermiques et sont composées d’une hyperkératose focale orthokératosique disposée en colonnes compactes avec une granuleuse peu développée.
Cancers associés : les néoplasies associées sont bronchiques, gastriques, mammaires, oesophagiennes, rénales, rectales et le mélanome.
Particularités : le caractère paranéoplasique de ces lésions serait inconstant (environ 35 % des cas) : certaines formes seraient héréditaires, et pour certains cette affection serait plus un marqueur cutané témoignant d’une prédisposition génétique à la survenue d’un cancer qu’un réel syndrome paranéoplasique.
6- Kératodermies palmoplantaires :
Les kératodermies palmoplantaires acquises ponctuées ou diffuses forment un groupe hétérogène.
Certaines kératodermies pourraient s’associer à des carcinomes pulmonaires ou gastriques mais l’exposition à certains carcinogènes comme l’arsenic pourrait à la fois expliquer la survenue de la kératodermie et d’un cancer.
Par ailleurs, la kératodermie du syndrome de Howel-Evans ou tylosis, génodermatose de transmission autosomique dominante, apparaît plus comme un marqueur de risque de survenue d’un carcinome oesophagien qu’une DPN.
Globalement, le caractère paranéoplasique de certaines kératodermies palmoplantaires reste controversé car la prévalence des lésions ne semble pas différente entre les patients atteints de néoplasie et la population générale.
7- Acrokératose paranéoplasique de Bazex :
Clinique : elle prédomine chez l’homme de plus de 50 ans, aux antécédents alcoolotabagiques.
Elle se caractérise par plusieurs stades évolutifs successifs.
L’éruption est initialement asymptomatique et de façon simultanée et symétrique surviennent sur les faces dorsales et palmaires des doigts, des orteils, sur l’arête nasale et sur l’hélix des plaques rouge violacé recouvertes d’éléments érythématosquameux adhérents, psoriasiformes, qui résistent aux traitements.
En l’absence de diagnostic et de traitement du cancer associé, les lésions ont une extension centripète aboutissant à une kératodermie palmoplantaire fissuraire, jaunâtre, respectant l’arche plantaire et la partie médiane des paumes.
Elle est associée à des dystrophies unguéales : ponctuations, hyperkératose sous-unguéale, paronychie.
Dans les formes tardives, le tableau est quasi érythrodermique par extension des lésions au cuir chevelu, aux membres, tronc, organes génitaux, et la néoplasie associée est souvent au-delà de toute sanction thérapeutique.
Histologie cutanée : elle n’est pas spécifique ; parakératose, acanthose, papillomatose, infiltrat lymphocytaire du derme superficiel.
L’IFD met parfois en évidence des dépôts d’IgG, A ou M et de C3 à la jonction dermoépidermique.
Cancers associés : dans la majorité des cas, la néoplasie associée est un épithélioma spinocellulaire des voies aérodigestives supérieures (cavum, sinus piriforme, cordes vocales, base de la langue), plus rarement l’oesophage ou les bronches.
Dans certaines observations, l’acrokératose se diagnostique dans un contexte de métastases ganglionnaires cervicales prévalentes d’un carcinome épidermoïde sans tumeur primitive identifiée.
Plus rarement, l’acrokératose de Bazex a été rapportée en association avec un carcinome vulvaire, un adénocarcinome gastrique, prostatique, une néoplasie thymique ou un myélome multiple, des lymphomes.
Signes associés : lichen pigmentaire, leucomélanodermie, prurit, vitiligo, pelade ou autres syndromes paranéoplasiques associés : ichtyose acquise, hypercalcémie, sécrétion inappropriée d’ADH.
Mécanisme : la participation de facteurs de croissance est probable : sécrétion de TGF a, EGF, IGF1 par les cellules tumorales agissant sur la kératinisation.
Un mécanisme immunologique est aussi possible, au vu des données de l’IFD et des dépôts IgM, A, G et de C3 au niveau de la membrane basale, par dépôt d’antigènes circulants (SCC-Ag ou squamous cell carcinoma antigen).
Traitement : c’est celui de la néoplasie sous-jacente.
Les rétinoïdes, en l’absence de traitement spécifique, ont été efficaces dans des observations ponctuelles.
8- Papillomatose cutanée :
Clinique : il s’agit d’une affection rare (moins de 25 cas), qui prédominerait chez l’homme d’âge moyen.
Elle se caractérise par l’éruption soudaine de lésions planes, papillomateuses ou verruqueuses indistinguables des papillomes et des verrues à human papilloma virus (HPV), en n’importe quel point du tégument (papillomatose cutanée) ou principalement au niveau des régions périorificielles : yeux, bouche et muqueuse buccale.
La papillomatose orale floride isolée est rarement paranéoplasique. Histologie : lésions non spécifiques à type d’hyperkératose, d’acanthose, de papillomatose sans les signes cytologiques évocateurs d’une infection des kératinocytes par des HPV (clarification cellulaire, densification des grains de kératohyaline).
La recherche de papillomavirus par technique d’hybridation reste négative.
Cancers associés : la néoplasie associée est souvent gastrique, plus rarement gynécologique, urologique ou pulmonaire.
Particularités : des associations privilégiées avec l’acanthosis nigricans paranéoplasique, avec le signe de Leser-Trélat, l’hypertrichose lanugineuse acquise, le vitiligo ou l’ichtyose acquise ont été rapportées.
L’apparition de la papillomatose serait tardive en cours d’évolution du néoplasme associé.
Mécanisme : la survenue des lésions serait en relation avec une sécrétion d’EGF par la tumeur.
9- Signe de Leser-Trélat ou éruption de kératoses séborrhéiques (KS) :
Clinique : le signe de Leser-Trélat est défini comme l’apparition brutale, « éruptive », de nombreuses KS ou comme une augmentation soudaine du nombre et de la taille des lésions de KS préexistantes, quelques mois avant la découverte d’une néoplasie sous-jacente chez un sujet âgé de 60 ans en moyenne.
La topographie préférentielle n’est pas différente de celle habituellement constatée pour ces kératoses : le tronc, les extrémités et la face.
Cancers associés : cette efflorescence de KS s’associerait dans plus de la moitié des cas à un adénocarcinome à point de départ gastrique (36 % des cas), mammaire, mais d’autres néoplasies ont été rapportées : lymphomes, carcinomes épidermoïdes bronchiques.
Mécanismes : la pathogénie est inconnue.
Certains auteurs tentent de rapprocher ce phénomène de l’acanthosis nigricans où la sécrétion de facteurs de croissance par la tumeur interviendrait.
Particularités : le signe de Leser-Trélat peut s’associer à un prurit (dans presque la moitié des cas) ou à d’autres lésions considérées comme paranéoplasiques, en particulier l’acanthosis nigricans (présent dans 35 % des cas).
La signification du signe de Leser-Trélat est très controversée pour diverses raisons :
– la régression des KS après le traitement du cancer est relativement inconstante, ce qui va à l’encontre des critères de Curth établissant un lien entre une dermatose et une néoplasie ;
– l’association entre KS et cancer peut être fortuite du fait de la grande fréquence des deux pathologies chez le sujet âgé ;
– il existe un « flou » dans la définition : le critère « multiple » des KS est peu fiable, de même que le caractère éruptif qui est très subjectif et difficile à authentifier chez une personne âgée.
Rampen a montré que le délai entre l’apparition des KS éruptives et le diagnostic de cancer tend à diminuer avec l’âge, ce qui peut correspondre à un biais de rappel ;
– d’autre part, des études cas-témoins n’ont pas montré d’augmentation du nombre de KS chez des sujets porteurs de diverses néoplasies.
E – AUTRES DERMATOSES PARANÉOPLASIQUES :
1- Hypertrichose lanugineuse acquise :
Clinique : appelée par certains auteurs « duvet malin », cette affection semble rare, avec moins de 50 cas décrits dans la littérature.
La majorité des sujets concernés sont de sexe féminin.
En quelques semaines se développe brutalement un duvet de poils sur les régions du corps habituellement glabre.
Les poils sont fins, doux et de couleur claire.
Le duvet prédomine en région cervicofaciale et concerne les joues, le nez, les oreilles, les paupières, le cou, mais il atteint aussi les extrémités et le tronc.
Ce duvet respecte les paumes, les plantes et les zones où se développe la pilosité terminale qui est sans changement.
L’intensité de la pilosité peut être très variable d’un patient à l’autre.
Histologie : l’étude histologique des lésions est plus en faveur d’une régression des follicules pileux existants vers le stade foetal que de l’apparition de nouveaux poils.
Les poils visibles prédominent en phase anagène et n’ont plus de gaine ni de médulla.
Cancers associés : la néoplasie associée est parfois de diagnostic tardif par rapport aux anomalies pilaires : il s’agit de carcinomes des voies biliaires, de carcinomes vésicaux, colorectaux, bronchopulmonaires, gynécologiques, souvent au stade métastatique, plus rarement de lymphomes.
Particularités : des anomalies sont parfois associées au niveau de la muqueuse buccale : glossite douloureuse avec une langue recouverte de papules oedémateuses ou langue fissuraire et leucokératosique.
Des associations à d’autres DPN semblent habituelles : acanthosis nigricans et pigmentation généralisée, ichtyose acquise, kératodermie palmoplantaire, kératoses séborrhéiques profuses, papillomatose cutanée floride.
La survenue rapide du tableau à l’âge adulte permet d’éliminer les formes congénitales et familiales d’hypertrichose lanugineuse qui s’associent à diverses anomalies dentaires et osseuses, voire à un retard mental.
Il n’existe par ailleurs aucun symptôme associé endocrinien, notamment aucun signe de virilisation, aucun signe métabolique (malnutrition) ou cutané (porphyrie), et aucune prise médicamenteuse favorisant l’hyperpilosité.
Mécanisme : le duvet serait secondaire à la production d’une substance capable de reproduire le lanugo physiologique qui se développe au cours de la vie foetale.
Les taux d’antigène carcinoembryonnaire (ACE) sont souvent augmentés au cours de cette DPN, mais on ne sait pas s’il joue un rôle ou s’il est seulement un simple « témoin » du cancer.
2- Pemphigus paranéoplasique :
Anhalt propose en 1990 le terme de pemphigus paranéoplasique pour décrire chez des patients porteurs de néoplasies le développement de lésions cutanées polymorphes associées à des anticorps dirigés contre certaines cibles moléculaires cutanées.
Clinique : le tableau clinique est hétérogène et associe des critères communs au pemphigus vulgaire, à la pemphigoïde bulleuse et à l’érythème polymorphe : maculopapules, cocardes, lésions lichénoïdes, érosions muqueuses pouvant toucher la cavité buccale, les conjonctives mais aussi l’oropharynx.
Les manifestations pulmonaires ne sont pas rares.
L’âge moyen est de 56 ans et le sexratio de 1:1. Histologie : l’association d’une acantholyse qui prédomine au niveau de la couche granuleuse, d’une dyskératose avec nécrose kératinocytaire et l’absence de spongiose à éosinophiles doivent faire évoquer le diagnostic.
Les lésions sont parfois aussi associées à une exocytose mononucléée sans vascularite.
L’IFD révèle des dépôts d’IgG et de complément au niveau de la substance intercellulaire et des dépôts de complément au niveau de la membrane basale (linéaire et/ou granulaire).
L’immunofluorescence indirecte sur oesophage de singe montre un profil d’IgG 1, 2, 3, 4 de prédominance lambda sur la substance intercellulaire.
L’utilisation de substrats tels que la vessie de rat est justifiée pour mettre en évidence des anticorps dirigés contre les desmoplakines présentes dans ces substrats alors que les desmogléines en sont absentes.
Les cibles antigéniques identifiées par immunotransfert (ou immunoblot) et immunoprécipitation sont des protéines de 190 kDa (périplakine), 210 et 250 kDa (respectivement desmoplakines II et I), 230 kDa (antigène majeur de la pemphigoïde bulleuse) ainsi que la desmogléine 3 et une protéine non identifiée de 170 kDa.
Cancers associés : ce pemphigus paranéoplasique est par définition associé à un cancer, le plus souvent une hémopathie maligne (Hodgkin, lymphome malin non hodgkinien, leucémie lymphoïde chronique, etc.).
Le plus souvent, le diagnostic de cancer est posé avant la survenue des lésions cutanées.
Des tableaux similaires ont cependant été décrits en association à diverses tumeurs bénignes (thymome, maladie de Castelman).
Mécanismes : le pemphigus paranéoplasique est une maladie autoimmune complexe pouvant provenir d’une levée de la tolérance envers certains antigènes impliqués dans les systèmes de jonction kératinocytaires.
Les anticorps produits par l’hôte en guise de défense contre la tumeur pourraient aussi être dirigés contre des épitopes cutanés.
La grande hétérogénéité clinique et paraclinique de cette entité a conduit certains auteurs à parler de « syndrome auto-immun multiorgane paranéoplasique ».
Certains auteurs suspectent aussi le rôle inducteur de certains traitements de la néoplasie comme l’interféron ou la radiothérapie par le déséquilibre immunitaire induit.
3- Autres dermatoses bulleuses observées au cours des syndromes paranéoplasiques :
* Pemphigoïde bulleuse :
Les observations sont ponctuelles et controversées.
Le jeune âge des sujets, la résistance à la corticothérapie par voie générale et l’absence d’anticorps circulants pourraient être évocateurs.
Cependant, les études contrôlées montrent une augmentation des cas de pemphigoïde bulleuse avec l’âge des sujets mais pas d’association significative avec les cancers.
* Dermatose à IgA linéaire :
De rares cas ont été décrits en association avec des affections lymphoprolifératives, plus rarement avec des cancers viscéraux.
Cependant, la majorité des observations restent idiopathiques ou en relation avec des prises médicamenteuses concomitantes.
* Autres :
Différentes observations ponctuelles de dermatoses bulleuses autoimmunes « intriquées ou inclassables » sont rapportées dans la littérature souvent au cours de lymphomes : elles ont l’avantage d’illustrer le phénomène de rupture de tolérance des lymphocytes B à différents antigènes épidermiques induits par le cancer, avec par exemple production d’autoanticorps dirigés contre les antigènes des desmosomes et des hémidesmosomes.
Conclusion :
Pour la pratique, on retiendra les points suivants.
Les dermatoses paranéoplasiques ne résultent pas d’une extension directe du cancer, mais leur présence est évocatrice d’une tumeur sousjacente dont le développement précède toujours les manifestations cutanées.
L’évolution de la dermatose est parallèle à celle du cancer avec régression lorsque le cancer est en rémission, et reprise évolutive en cas de récidive de la néoplasie.
Les dermatoses spécifiquement associées au cancer sont rares mais très évocatrices d’une tumeur profonde qu’il faudra rechercher de principe.







")

")
")
{kind=link}