



















Introduction :
Le terme de lupus a été initialement utilisé à la fin du Moyen Âge pour décrire des lésions cutanées mutilantes du visage de causes variées.
Actuellement, le terme de lupus érythémateux désigne un ensemble d’affections formant un spectre continu allant d’une lésion cutanée isolée à une maladie multiviscérale grave dans le cadre d’un lupus érythémateux aigu disséminé (LEAD), aussi appelé lupus systémique.
Les nombreuses manifestations cutanéomuqueuses observées dans ces affections ont une grande valeur diagnostique et parfois pronostique.
Aussi est-il fondamental d’en faire un diagnostic précis.
Schématiquement, ces manifestations peuvent être classées en trois groupes : les lésions lupiques caractérisées par une atteinte de l’interface dermoépidermique, les lésions vasculaires, les manifestations non lupiques et non vasculaires.
Lésions lupiques :
 Il n’existe pas de définition précise des lésions lupiques.
Il n’existe pas de définition précise des lésions lupiques.
Arbitrairement, dans un but de simplification de classement des lésions dermatologiques observées au cours du lupus, nous avons considéré que les lésions lupiques étaient caractérisées par une atteinte de l’interface de la jonction dermoépidermique, atteinte non spécifique puisque observée dans d’autres maladies telles que la dermatomyosite.
Le diagnostic de lésion lupique repose en fait sur un faisceau d’arguments prenant en compte l’aspect clinique des lésions dermatologiques, leur topographie, leur évolution, une histologie compatible, les résultats de l’immunofluorescence cutanée directe, le contexte clinique et immunologique.
A – NOSOLOGIE. ÉPIDÉMIOLOGIE. FACTEURS DE RISQUE :
1- Lupus érythémateux aigu :
Le lupus érythémateux aigu (LEA) est observé essentiellement chez la femme, avec un sex-ratio femmes/hommes de 9/1.
Il serait plus souvent observé en cas de début précoce du lupus.
Mieux visible sur des malades à carnation claire, il est probablement sous-évalué chez les malades à carnation très foncée, mais aussi peut-être moins fréquent en raison de leur protection naturelle aux ultraviolets (UV).
La multitude des études génétiques chez les malades avec un LEAD contraste avec l’absence d’études recherchant une relation entre certains haplotypes human leukocyte antigen (HLA) et le LEA.
2- Lupus érythémateux subaigu :
Autrefois dénommé sous des appellations diverses, le lupus érythémateux subaigu (LES) a été individualisé en 1977 par Gilliam.
Il atteint préférentiellement les femmes (70 %) d’origine caucasienne (85 %), surtout de la cinquième décennie.
Le LES serait préférentiellement associé aux haplotypes HLA-B8, DR3.
Les malades ayant également une symptomatologie de Gougerot- Sjögren auraient plus souvent les haplotypes B8, DR3, DRW6, DQ1/2 et DRW52, ces haplotypes favorisant une forte production d’anticorps anti-Ro (SS-A).
3- Lupus érythémateux chronique :
Le lupus érythémateux chronique (LEC) regroupe le lupus discoïde, le lupus tumidus, le lupus à type d’engelures et la panniculite ou lupus profond qui comporte inconstamment une atteinte de la jonction dermoépidermique.
Le terme de LEC est souvent confondu avec celui de lupus discoïde, le lupus tumidus et le lupus à type d’engelures étant alors considérés comme des formes cliniques de lupus discoïde.
La panniculite est à part.
Il n’existe aucune étude épidémiologique concernant la prévalence du LEC.
Certains auteurs le considèrent deux à trois fois plus fréquent que le lupus systémique.
À l’inverse, d’autres le considèrent sept fois moins fréquent.
La prévalence du LEAD ayant été évaluée entre 14,6 et 50,8/100 000 suivant les régions, la prévalence du LEC serait comprise entre 2 et 100/100 000, l’ampleur de cet intervalle témoignant de l’absence de données épidémiologiques fiables.
Le LEC débute souvent entre 20 et 40 ans, mais peut également atteindre les âge extrêmes de la vie.
La prédominance féminine est moins nette que dans les autres formes, le sex-ratio femmes/hommes variant de 3/2 à 3/1.
Toutes les races sont atteintes, certaines études récentes aux États- Unis étant en faveur d’une prévalence plus élevée chez les Noirs.
Chez les malades avec LEC, a été démontrée une augmentation significative des antigènes HLA B7, B8, CW7, DR2, DR3, DQW1 et une diminution de l’HLA-A2.
La présence combinée de CW7, DR3, DQW1 et de B7, Cw7 et DR3 conférerait un risque maximal (X 7,4) de présenter un LEC. Un phénomène de Koebner explique certaines localisations des lésions de LEC sur des zones traumatisées ou sur des cicatrices préexistantes.
4- Photosensibilité :
La fréquence du déclenchement par l’exposition solaire des lésions lupiques est très difficile à évaluer.
En effet, les données de l’interrogatoire des malades ne sont pas toujours fiables, du fait notamment du caractère retardé de l’apparition des lésions lupiques par rapport à l’exposition solaire et de la prise de conscience individuelle très variable des différentes expositions solaires possibles.
Les résultats des tests de provocation varient en fonction des techniques utilisées et de la population étudiée en raison de facteurs génétiques.
Grossièrement, le déclenchement des lésions cutanées par l’exposition solaire est noté chez 25 à 30 % des malades ayant un LEA, 65 à 80 % des malades ayant un LES et 30 à 40 % des malades ayant un LEC, les rayonnements nocifs étant surtout les UVB et à un moindre degré les UVA et le visible.
Cette photosensibilité des lésions lupiques serait plus élevée chez les malades de moins de 40 ans que chez les sujets plus âgés.
Sa corrélation avec la présence sérique d’anticorps anti-Ro n’a pas été mise en évidence dans toutes les études.
La radiothérapie par rayons X peut être également un facteur déclenchant de lésions de lupus cutané, notamment de LES.
5- Lupus induits :
Ils représentent environ 10 % des lupus systémiques. Parmi la multitude de médicaments inducteurs possibles, les médicaments le plus souvent incriminés sont l’hydralazine, la procaïnamide, l’isoniazide et les bêtabloquants.
Habituellement, ces lupus induits ont peu de manifestations cutanées, rénales ou neurologiques.
Il faut cependant signaler qu’une éruption cutanée (de type LEA) a été signalée dans 18 % des lupus induits par la procaïnamide et 26 % des lupus induits par l’hydralazine.
Biologiquement, ces lupus induits sont caractérisés par un taux élevé d’anticorps antinucléaires contrastant avec l’absence habituelle d’anticorps antiacide désoxyribonucléique (ADN) natif et d’hypocomplémentémie.
Un certain nombre de médicaments ont été associés à la survenue ou à l’aggravation de lésions de LES.
La famille médicamenteuse la plus incriminée est celle des diurétiques thiazidiques.
Les autres médicaments ont été occasionnellement rapportés : procaïnamide, D-pénicillamine, glyburide (sulfonylurée), oxyprénolol, griséofulvine, piroxicam, naproxène, spironolactone, diltiazem, cilazapril, interféron (IFN)-alpha, cinnarizine. Il n’est pas de règle de rechercher une cause médicamenteuse dans les LEC.
Le problème des contraceptifs oestroprogestatifs est différent.
Ils ne sont pas réellement inducteurs mais peuvent déclencher des poussées systémiques de lupus.
Leur responsabilité dans l’aggravation de LES et LEC n’a pas été clairement établie.
Il paraît cependant logique de les éviter en cas de lupus cutanés résistants à l’hydroxychloroquine et/ou accompagnés d’anomalies immunologiques sériques.
Quant au traitement substitutif de la ménopause, aucune donnée actuelle ne permet de le contre-indiquer dans les lupus uniquement cutanés.
Il est parfois prescrit dans les lupus systémiques stabilisés.
6- Déficits immunologiques associés :
Les déficits génétiques en fractions du complément ainsi que le déficit congénital en inhibiteur de la C1-estérase ont été surtout rapportés en association avec des LES et des LEA, mais aussi avec des LEC.
Les malades atteints de granulomatose septique ou leurs mères peuvent avoir des lésions proches de celles du lupus discoïde.
L’immunofluorescence directe y est cependant généralement négative.
Il s’y associe fréquemment des aphtes buccaux. Un cas a été décrit associé avec un LEAD.
B – PHYSIOPATHOLOGIE :
La physiopathologie des lupus cutanés reste un puzzle dont il manque encore de nombreuses pièces.
Les lupus cutanés résultent vraisemblablement, comme le lupus systémique, d’interactions entre des gènes de susceptibilité et des facteurs d’environnement, ayant pour conséquence une réponse immune anormale comportant une hyperréactivité lymphocytaire T et B qui n’est pas réprimée par les circuits habituels d’immunorégulation.
Cette réponse immunitaire est essentiellement localisée au niveau de l’interface dermoépidermique, ce qui suggère l’existence de cibles antigéniques situées à la surface des kératinocytes de la couche basale de l’épiderme.
Les facteurs d’environnement qui déclenchent une poussée cutanée de la maladie sont pour la plupart inconnus, à l’exception des UVB et à un moindre degré des UVA.
Récemment, des données ont précisé les mécanismes par lesquels l’apoptose kératinocytaire induite par les UV pourrait déclencher des poussées, voire l’apparition de la maladie.
L’apoptose est un mécanisme génétiquement programmé par lequel une cellule meurt sans entraîner de nécrose, c’est-à-dire sans réaction inflammatoire.
Très souvent, ce mécanisme est déclenché par la stimulation d’un récepteur membranaire, appelé Fas (Apo-1, appartenant à la superfamille des récepteurs au tumor necrosis factor [TNF]), par un Fas-ligand présent notamment sur les cellules du système immunitaire, essentiellement les lymphocytes T.
Les kératinocytes sont capables d’exprimer cette molécule Fas, notamment après irradiation UV. Il a récemment été montré que les kératinocytes des patients lupiques pourraient ainsi être beaucoup plus sensibles à l’apoptose induite par les UV.
D’autre part, la fréquence élevée de lupus cutanés associés à un déficit en C1q et le rôle protecteur potentiel du C1q en situation physiologique sur l’apoptose kératinocytaire ont reçu récemment une explication à l’échelon moléculaire.
Les déficits en fractions du complément réduisent globalement la clairance des complexes antigènes-anticorps de l’organisme, mais cette donnée physiopathologique essentielle n’avait jusqu’à présent jamais été reliée directement aux poussées cutanées.
Il semble actuellement que la voie classique du complément puisse maintenir la tolérance immune.
Les kératinocytes entrant en apoptose se mettent à exprimer à leur surface des vésicules contenant des autoantigènes intracellulaires qui étaient jusqu’à présent masqués au système immunitaire.
La mauvaise élimination de ces autoantigènes favoriserait leur présentation aux lymphocytes T autoréactifs et la rupture de la tolérance sur un terrain génétique prédisposé.
En revanche, en situation normale, il semble bien que les vésicules apoptotiques ainsi formées à la surface des kératinocytes acquièrent la capacité de lier spécifiquement le C1q en l’absence même de la constitution de complexes immuns.
Ainsi, le C1q pourrait favoriser une clairance des kératinocytes apoptotiques indépendamment de la présence d’anticorps, et alors prévenir l’immunisation par des autoantigènes d’origine cutanée.
Les lésions cutanées de lupus discoïde expriment des cytokines lymphocytaires, avec un profil de sécrétion aberrant et probablement unique en physiopathologie.
Une synthèse abondante d’interleukine (IL) 5 est notamment présente dans les trois formes cliniques, ce qui suggère une réaction immunitaire cutanée de type Th2.
Ce type de réaction oriente la réponse vers l’immunité humorale, ce qui est tout à fait cohérent avec les données obtenues dans le LEAD.
En revanche, l’IL4 ne semble pas particulièrement présente dans les peaux lupiques, sans doute car il existe dans le même temps une synthèse accrue d’IFN-gamma antagoniste de la production d’IL4.
Ainsi, le profil de sécrétion des cytokines avec présence d’IFN-gamma (cytokine Th1) augmenterait l’expression de molécules d’adhésion comme l’ICAM-1 à la surface des kératinocytes, ce qui contribuerait à faciliter le recrutement de cellules pro-inflammatoires.
D’autre part, les cytokines Th2 (IL5 essentiellement) favorisent l’apparition des autoanticorps.
Enfin, l’IL2 ne semble pas transcrite dans les lésions lupiques cutanées, ce qui confirme que la prolifération lymphocytaire Th1 n’est qu’un événement accessoire dans l’apparition des lésions.
La régulation aberrante de l’IFN-gamma sans IL2 est similaire à celle observée en réponse à l’apport exogène d’IFN-gamma.
Enfin, l’irradiation UV peut in vivo faire synthétiser de l’IL10 par les kératinocytes humains ; cette notion, longtemps admise uniquement dans les modèles murins, a été récemment mise en évidence.
Cette cytokine de type Th2 est largement impliquée dans l’activation lymphocytaire B et la synthèse d’autoanticorps.
La démonstration de son expression accrue dans les peaux lupiques est fortement suspectée mais n’a pas encore été formellement démontrée.
De plus, la synthèse des cytokines pro-inflammatoires précédemment citées est parfois liée à l’expression dans les kératinocytes d’une enzyme, la monoxyde d’azote (NO)-synthase inductible, qui joue le rôle d’un second messager pro-inflammatoire.
Cette enzyme est en effet exprimée dans des lésions lupiques, aussi bien aiguës que chroniques.
Les UV étant eux-mêmes capables d’induire l’expression de la NO-synthase dans les kératinocytes irradiés, ces résultats apportent une explication supplémentaire à l’inflammation cutanée liée à l’irradiation UV dans la peau ; le NO ainsi produit peut aussi être à l’origine du déclenchement du processus apoptotique.
C – ASPECTS CLINIQUES :
1- Lupus érythémateux aigu :
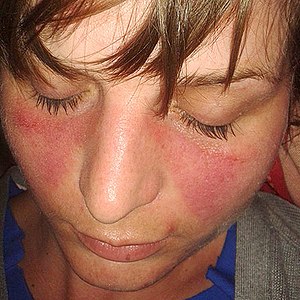
Il est caractérisé cliniquement par son aspect érythémateux, plus ou moins oedémateux ou squameux, voire papuleux.
Dans la forme localisée, il est situé principalement sur les joues et le nez, en vespertilio ou en « loup », respectant relativement les sillons nasogéniens, s’étendant souvent sur le front, les orbites, le cou, dans la zone du décolleté.
L’oedème, parfois important, peut gêner l’ouverture des yeux.
Dans la forme diffuse, il prédomine généralement sur les zones photoexposées, réalisant une éruption morbilliforme, papuleuse, eczématiforme ou bulleuse.
Sur le dos des mains, les lésions lupiques atteignent surtout les zones interarticulaires qui, à l’inverse, sont respectées dans la dermatomyosite.
Dans les formes bulleuses du lupus suraigu existent de vastes décollements, survenant toujours en zones érythémateuses lupiques.
Les lésions buccales de lupus aigu sont érosives, localisées préférentiellement sur les gencives, le palais, les joues ou les lèvres, tantôt bien supportées, tantôt très douloureuses, gênant l’alimentation.
L’atteinte génitale est beaucoup plus rare, généralement associée à une atteinte buccale.
Toutes ces lésions régressent rapidement sans cicatrice en dehors d’une possible hyperpigmentation séquellaire chez le sujet noir.
Le diagnostic différentiel se pose surtout avec la rosacée qui comporte des télangiectasies et des pustules, avec une dermite séborrhéique localisée principalement dans les plis nasogéniens, avec une dermatomyosite prédominant au visage sur les paupières supérieures, de couleur lilacée, et aux mains sur les zones articulaires.
Les formes disséminées évoquent parfois un eczéma, une éruption virale ou toxidermique.
Les formes suraiguës peuvent faire discuter une nécrolyse épidermique toxique.
Les érosions buccales du lupus aigu sont généralement moins étendues que celles du syndrome de Stevens-Johnson.
L’atteinte génitale lupique est plus rare et généralement plus limitée.
2- Lupus érythémateux subaigu :
Cliniquement, le LES se manifeste initialement par des lésions maculeuses érythémateuses ou papuleuses évoluant soit vers une forme annulaire, soit vers une forme psoriasiforme.
Dans la forme annulaire, les lésions ont des contours polycycliques à bordure érythématosquameuse ou vésiculocroûteuse avec un centre hypopigmenté grisâtre parfois couvert de télangiectasies. Rarement, elles peuvent prendre un aspect d’érythème polymorphe (syndrome de Rowell).
Dans la forme psoriasiforme, les lésions sont papulosquameuses, psoriasiformes ou pityriasiformes, pouvant confluer pour réaliser une forme profuse, voire une érythrodermie exfoliative.
Les deux formes peuvent être associées chez un même malade.
Quelle que soit la forme, l’atteinte est superficielle, sans kératose folliculaire visible ni squame adhérente.
Les lésions ont une topographie évocatrice du fait d’une distribution prédominante sur les zones photoexposées avec une atteinte grossièrement symétrique du visage, du cou, du décolleté, des épaules, de la face d’extension des bras, du dos des mains.
L’extension sur le tronc est possible avec respect fréquent de la face interne des membres supérieurs, des aisselles et des flancs.
L’atteinte des membres inférieurs est rare.
La régression des lésions est plus ou moins rapide, sans atrophie cicatricielle mais avec des troubles pigmentaires (hypo- ou hyperpigmentation) et des télangiectasies séquellaires.
Le diagnostic peut hésiter avec une dermatophytie, un eczéma annulaire, un érythème polymorphe, un psoriasis, un pityriasis rosé de Gibert, une toxidermie.
L’examen anatomopathologique avec immunofluorescence directe en zone lésionnelle permet d’éliminer la majorité de ces diagnostics, excepté l’érythème polymorphe en cas de syndrome de Rowell ou une toxidermie en cas de lésions diffuses.
Dans ces derniers cas, c’est le contexte clinique qui oriente vers le diagnostic de LES.
3- Lupus érythémateux chronique :
Il regroupe le lupus discoïde, le lupus tumidus, le lupus à type d’engelures ou lupus pernio, le lupus profond ou panniculite lupique.
* Lupus discoïde :
Dans sa forme classique, le lupus discoïde réalise des plaques bien limitées associant trois lésions élémentaires :
– érythème de type congestif surtout net en bordure, parcouru de fines télangiectasies ;
– squames plus ou moins épaisses s’enfonçant en « clous » dans les orifices folliculaires pouvant donner un aspect de piqueté blanc, râpeux au toucher ;
– atrophie cicatricielle prédominant au centre des lésions souvent dépigmenté, parfois tatoué de télangiectasies et de taches pigmentées.
Les lésions, souvent multiples et symétriques, sont surtout localisées sur les zones photoexposées, notamment au visage sur l’arête du nez, les pommettes, avec parfois une disposition en « aile de papillon », les régions temporales et l’ourlet des oreilles.
Les zones non exposées sont en fait souvent atteintes, en particulier les sourcils, les paupières ou le cuir chevelu.
Ainsi, des plaques du cuir chevelu existent-elles dans 60 % des cas, isolées dans 10 %, laissant après guérison une alopécie cicatricielle définitive avec un aspect de pseudopelade.
Ailleurs les lésions sont plus diffuses, atteignant le tronc et les membres et on parle alors de lupus discoïde disséminé.
Sur les membres, les lésions sont observées préférentiellement sur les zones traumatisées comme les coudes ou sur les extrémités.
L’atteinte palmoplantaire est souvent érosive, très douloureuse, particulièrement résistante aux traitements, invalidante sur le plan fonctionnel, gênant la marche en cas de lésions plantaires et empêchant toute activité manuelle en cas de lésions palmaires.
L’atteinte unguéale est rare, à l’origine de dystrophies pseudolichéniennes. Des lésions muqueuses, essentiellement buccales, seraient présentes dans 25 % des cas.
Initialement, il s’agit de lésions érythémateuses évoluant vers un aspect lichénien avec des zones blanches isolées ou entourant des zones érythémateuses ou érosives en « rayons de miel ».
Les demi-muqueuses des lèvres, la face interne des joues et le palais sont le plus souvent atteints, alors que l’atteinte linguale est plus rare.
L’évolution vers un carcinome spinocellulaire est possible.
L’atteinte des autres muqueuses, notamment conjonctivale, nasale ou génitale, est rare.
Différentes formes cliniques existent selon la prédominance ou la répartition des lésions élémentaires : lupus crétacé très hyperkératosique, lupus comédonien avec de nombreux comédons ouverts, lupus folliculaire notamment des coudes, fréquent chez les Asiatiques, lupus télangiectasique, formes érythémateuses, très difficiles à distinguer du lupus aigu, formes infiltrées nodulaires avec un centre déprimé kératosique sur les mains, lupus atrophique avec des lésions cicatricielles vermoulues de la zone péribuccale.
* Lupus tumidus :
Il réalise un ou plusieurs placards nettement saillants, arrondis ou ovalaires, de teinte rouge violacé, à bords nets comme tracés au compas, de consistance oedémateuse, sans hyperkératose folliculaire visible à l’oeil nu.
Certaines lésions sont déprimées en leur centre.
Les lésions sont principalement localisées au visage, parfois sur le tronc.
Elles disparaissent habituellement sans cicatrice.
Le diagnostic différentiel se pose essentiellement avec le LES et les infiltrats lymphocytaires bénins cutanés, en particulier de Jessner-Kanoff.
* Lupus à type d’engelures :
Il est caractérisé par sa localisation (extrémités des doigts et des orteils, oreilles, nez, mollets, talons, coudes, genoux), son évolution souvent saisonnière aggravée par le froid, son aspect clinique avec des lésions violacées souvent ulcérées, prurigineuses ou douloureuses.
Cliniquement, il peut être confondu avec des engelures, une sarcoïdose ou plus fréquemment avec des lésions de vasculite. L’histologie est voisine de celle du lupus discoïde.
Du fait de ces difficultés diagnostiques, des critères diagnostiques ont été proposés, comportant deux critères majeurs (lésions des extrémités induites par l’exposition au froid ou une diminution de la température et présence de lésions évocatrices de lupus en histologie avec immunofluorescence directe) et trois critères mineurs (coexistence d’un lupus systémique ou de lésions de lupus discoïde, réponse à un traitement des lupus cutanés et absence de cryoglobuline, cryofibrinogène ou d’agglutinines froides).
Les frontières entre le lupus à type d’engelures et les formes distales de lupus discoïde sont assez floues, les lésions étant classées lupus à type d’engelures lorsqu’elles sont aggravées par le froid et lupus discoïde lorsqu’elles n’ont pas de variations saisonnières.
* Panniculite lupique (ou lupus érythémateux profond ou maladie de Kaposi-Irgang) :
Elle est classiquement considérée comme une forme de LEC.
En fait, il n’existe qu’inconstamment une atteinte de l’interface dermoépidermique d’où la possibilité de classer également ces lésions comme non lupiques et non vasculaires.
La panniculite se manifeste par des nodules ou des plaques infiltrées de taille variable, parfois douloureuses.
La peau en regard est normale ou érythémateuse, parfois siège de lésions de lupus discoïde.
Les lésions s’ulcèrent dans 30 % des cas.
Les dépôts calciques sont inconstants.
L’évolution se fait vers une lipoatrophie cicatricielle permettant un diagnostic rétrospectif.
Il n’y a pas de fièvre. Les lésions siègent préférentiellement sur le tiers supérieur des bras, les joues ou les cuisses.
Plus rarement sont atteints les seins, avec un aspect pouvant simuler à la mammographie un carcinome inflammatoire, ou en régions abdominale, périoculaire ou parotidienne.
Le diagnostic différentiel se pose cliniquement avec les vasculites nodulaires ou les autres panniculites : panniculite factice, panniculite histiocytaire cytophagique, panniculites de Weber-Christian ou pancréatique, habituellement fébriles.
L’examen histologique permet généralement de faire le diagnostic.
4- Association des différents types de lupus cutanés :
Les différents types de lupus cutanés peuvent être associés chez un même malade.
Ainsi, sur 191 malades avec un lupus cutané, 68 % n’avaient qu’un seul type de lésion, 29 % en avaient deux et 3 % en avaient trois. Les lésions de LEA sont associées aussi bien au LES qu’au LEC.
Vingt pour cent environ des malades avec des lésions de LES ont ou auront également, au cours de leur évolution, des lésions de lupus discoïde.
Dans 70 % des cas, la panniculite est associée à des lésions de lupus discoïde en regard des lésions ou à distance.
D – ASPECTS HISTOLOGIQUES :
L’examen anatomopathologique d’une lésion cutanée lupique révèle dans les trois formes de lupus cutané des lésions épidermiques et dermiques avec une hyperkératose, une atrophie du corps muqueux, des lésions de dégénérescence des kératinocytes basaux, un épaississement de la membrane basale et un infiltrat lymphocytaire dermique composé essentiellement de lymphocytes CD4.
Des variations importantes existent suivant chaque forme de lupus.
Ainsi, dans le LEA, l’hyperkératose est peu importante ; l’infiltrat mononucléé est discret, surtout périvasculaire ; il existe un oedème du derme superficiel ; la dégénérescence des kératinocytes est souvent intense, plus étendue, notamment dans les formes sévères.
Dans le LES, les lésions de dégénérescence des kératinocytes sont également parfois très intenses ; l’hyperkératose est discrète ; l’infiltrat est peu abondant, périvasculaire et périannexiel.
Dans le lupus discoïde, l’hyperkératose est marquée, de type orthokératosique, formant des bouchons cornés dans les orifices folliculaires ; l’infiltrat dermique est plus important, périannexiel pouvant s’étendre dans le derme profond d’où l’évolution cicatricielle.
Dans le lupus tumidus existent une hyperkératose folliculaire minime, une discrète vacuolisation des kératinocytes basaux et un infiltrat inflammatoire abondant.
L’aspect histologique du lupus à type d’engelures est proche de celui du lupus discoïde avec une hyperkératose moins importante.
Au cours de la panniculite existe inconstamment un aspect de lupus discoïde dans le derme et l’épiderme.
Plus en profondeur, dans l’hypoderme, sont notés un infiltrat lobulaire composé de lymphocytes, de plasmocytes et d’histiocytes, des débris nucléaires, des dépôts fibrinoïdes, une nécrose hyaline des adipocytes, une hyalinisation des septa. Des foyers de calcification sont parfois présents.
L’étude en immunofluorescence directe d’une lésion lupique met en évidence des dépôts granulaires d’immunoglobulines (IgG, A ou M) et/ou de complément (C1q, C3) à la jonction dermoépidermique dans 80 à 90 % des cas de LEA et lupus discoïde, 70 % des cas de panniculite lupique et 60 % des cas de LES.
Ces dépôts ne sont pas spécifiques de la maladie lupique ; ils peuvent être observés dans certaines rosacées, les dermatomyosites et chez 20 % des sujets normaux en peau saine exposée.
Dans le LES a été décrite une fluorescence en poussières épidermiques dont la fréquence de positivité semble varier beaucoup en fonction de problèmes techniques.
Dans une étude récente, elle a été mise en évidence chez 31 % des malades avec LES, mais aussi chez des malades avec LEAD, lupus discoïde, connectivite mixte, syndrome de Gougerot-Sjögren ou des dermatoses variées. Seuls 36 % des 56 malades avec cette fluorescence avaient des anticorps anti-Ro (SS-A).
Histologiquement, le diagnostic est très difficile, voire impossible, entre une lésion de LEA et une lésion de dermatomyosite qui peut comporter également une bande lupique en immunofluorescence directe.
En cas de nécrose extensive des kératinocytes d’une lésion de LEA ou de LES, l’aspect histologique peut être proche de celui d’une toxidermie, surtout en cas de négativité de l’immunofluorescence directe.
E – ASSOCIATION AU LUPUS ÉRYTHÉMATEUX AIGU DISSÉMINÉ :
1- Définition. Prévalence de l’association :
Le LEAD est généralement défini par la présence d’au moins quatre critères sur les onze établis par l’American Rheumatology
Association (ARA) en 1982, deux de ces critères ayant été modifiés en 1997.
Or, ces critères ont été élaborés initialement dans un but de classification des maladies rhumatologiques ; ils ont été secondairement déviés de leur vocation initiale et utilisés en pratique comme critères diagnostiques de LEAD dans toutes les spécialités.
La présence de quatre critères dermatologiques conduit à classer abusivement des malades avec une atteinte cutanée isolée associée à quelques anomalies biologiques dans le groupe des LEAD alors qu’ils n’ont en fait aucune manifestation systémique.
Ce classement n’a aucune conséquence pratique puisque le choix du traitement dépend uniquement de l’existence et de la gravité des atteintes viscérales actuelles et non du nombre de critères de l’ARA comptabilisés depuis le début de la maladie.
Tous les types de lupus cutanés peuvent être associés à un LEAD.
Toutefois, la fréquence de cette association est très variable selon le type de lupus.
Ainsi, plus de 90 % des malades avec un LEA ont ou auront un LEAD, les lésions dermatologiques étant révélatrices dans 25 % des cas.
À l’inverse, 16 à 61 % des LEAD ont des lésions de LEA.
Celles-ci accompagnent très souvent les poussées de lupus systémique qu’elles doivent faire rechercher systématiquement.
Dans notre expérience, les ulcérations muqueuses sont fréquemment associées à une atteinte rénale évolutive.
Plus de 50 % des malades avec des lésions de LES ont un LEAD selon les critères de l’ARA.
En fait, la large majorité des malades avec LES n’ont pas d’atteinte systémique justifiant une corticothérapie générale.
Les atteintes viscérales graves, en particulier rénales ou neurologiques, seraient présentes dans près de 10 % des cas.
Ces dernières seraient surtout observées chez l’homme, avec un aspect papulosquameux psoriasiforme du LES.
À l’opposé, suivant les séries, 7 à 21% des malades avec un LEAD ont des lésions de LES.
Quinze à 20 % des malades avec LEAD ont des lésions cutanées de lupus discoïde.
À l’inverse, 10 à 20 % des malades avec un lupus discoïde ont ou auront un LEAD.
Environ 8 % des malades avec lupus discoïde initialement isolé évolueront vers un LEAD, le plus souvent après plusieurs années.
Il n’existe pas de critère prédictif formel de cette évolution ; dans certaines séries cependant, le caractère disséminé des lésions cutanées, leur aggravation en période prémenstruelle ou pendant la grossesse étaient plus souvent associés à une évolution vers un LEAD.
Près de 40 % des malades avec une panniculite lupique ont un LEAD.
À l’inverse, un aspect de panniculite n’est noté que chez 2 à 3% des LEAD.
2- En pratique :
Ces données justifient, devant tout lupus cutané, la recherche systématique par l’interrogatoire et l’examen clinique de manifestations suggestives de LEAD, ainsi que certains examens paracliniques : numération formule sanguine, vitesse de sédimentation, recherche d’anticorps antinucléaires et, en cas de positivité d’anticorps anti-ADN natif, d’anticorps anti-Ro (SS-A), d’anticorps anti-ECT, dosage du complément hémolytique total et de ses fractions C3, C4, examen cytobactériologique urinaire, protéinurie des 24 heures, électrocardiogramme.
Des anticorps antinucléaires sont présents dans environ 20 % des lupus discoïdes et 70 % des LES.
Les anticorps anti-SSA sont surtout observés dans les LES où leur prévalence varie de 60 à 80 % selon les séries et les techniques utilisées.
Ces anticorps sont parfois présents alors que le dépistage global des anticorps antinucléaires est négatif.
Dans le LEAD, des anticorps antinucléaires sont présents dans 98 % des cas et des anticorps anti-SSA dans 25 à 50 % des cas.
La pratique d’une immunofluorescence cutanée directe en peau saine a un intérêt diagnostique en l’absence de lésion dermatologique biopsiable.
En effet, des dépôts d’Ig et/ou de complément (bande lupique) sont présents à la jonction dermoépidermique dans 36 à 90 % des cas de LEAD.
Cette bande lupique est plus souvent observée en zone exposée qu’en zone non exposée, sa spécificité étant moindre en zone exposée.
Elle n’a pas de valeur prédictive d’une atteinte viscérale.
Au terme de ce bilan, le malade est situé dans le vaste spectre de la maladie lupique.
La possibilité d’évolution systémique justifie une surveillance clinique et biologique prolongée, la répétition du bilan biologique étant généralement inutile en l’absence de modification du contexte clinique.
F – AUTRES ASSOCIATIONS :
Tous les types de lupus cutanés peuvent être associés à diverses connectivites, la fréquence de ces associations étant généralement inconnue. De 3 à 20% des malades avec un LES développent un syndrome de Gougerot-Sjögren, ces pourcentages étant plus élevés chez les sujets âgés de plus de 55 ans.
Au LES et au syndrome sec s’ajoutent alors fréquemment une vascularite cutanée, une atteinte neurologique centrale et périphérique et un syndrome interstitiel pulmonaire.
Les éruptions annulaires décrites chez les Japonais avec syndrome de Gougerot-Sjögren primaire et anticorps anti-Ro (SS-A) ou anti-
La (SS-B) sont très proches du LES.
Elles en différeraient par un aspect clinique plus oedémateux avec en histologie absence des altérations de la jonction dermoépidermique et la possibilité de dépôts de mucine.
L’immunofluorescence directe est alors souvent négative.
Si un tiers environ des malades avec LES ont un facteur rhumatoïde dans leur sérum, environ 10 % des malades avec polyarthrite rhumatoïde ont des anticorps anti-Ro (SS-A).
L’association polyarthrite rhumatoïde et LES est pourtant rare, peu de cas étant rapportés dans la littérature.
Enfin, certaines lésions de LES ont parfois été considérées comme paranéoplasiques du fait de l’association dans de rares observations à des néoplasies diverses : cancer du poumon, cancer du sein, adénocarcinome utérin, cancer gastrique, mélanome malin, maladie de Hodgkin.
G – LUPUS NÉONATAL :
Sa prévalence est inconnue, probablement inférieure à 1/20 000 naissances. Une atteinte cutanée est présente dans approximativement la moitié des cas.
Le sex-ratio femmes/hommes est de 3/1 en cas d’atteinte cutanée.
Les lésions, rarement présentes dès la naissance, apparaissent habituellement dans les premières semaines de vie.
Il s’agit de plaques érythématosquameuses, arrondies et polycycliques, très proches cliniquement et histologiquement du LES, fréquemment hypopigmentées, notamment sur peau noire.
Les lésions sont principalement localisées sur la tête et les zones photoexposées, tout le tégument pouvant être atteint.
Elles disparaissent après une ou plusieurs poussées successives évoluant sur des semaines ou des mois, avec possibilité de télangiectasies ou de troubles pigmentaires séquellaires.
D’autres manifestations ont été décrites : lupus discoïde sans hyperkératose folliculaire marquée ni atrophie ; érosions buccales ; alopécie ; éruptions érythématosquameuses, purpuriques, bulleuses ; lésions télangiectasiques ; papules angiomateuses ; panniculite.
Les autres atteintes sont dominées par les troubles de la conduction cardiaque, associés à l’atteinte cutanée dans 11 % des cas seulement.
Des anticorps anti-Ro sont généralement présents chez la mère et l’enfant, plus rarement des anticorps anti-La ou des anticorps anti-U1RNP parfois isolés.
Récemment a été mise en évidence la fréquence des anticorps anticalréticuline.
Seulement 1 % environ des bébés de mères avec anticorps antiRo ont un lupus néonatal ; ce pourcentage s’élève à 15 % lorsqu’un premier enfant a déjà été atteint.
H – TRAITEMENT DES LUPUS CUTANÉS :
1- Photoprotection :
Quel que soit le type de lupus cutané, une protection solaire est indispensable.
Aussi l’utilisation de la photoprotection externe doitelle être large et systématique sur les zones découvertes dans la vie courante.
Les écrans solaires associent des molécules filtrantes sélectives des UVB et/ou des UVA et des poudres minérales formant un écran dispersées dans un excipient.
Les indices de protection contre les UVB, les UVA et/ou le visible correspondent à l’augmentation plus ou moins importante de la dose érythémateuse minimale qu’ils confèrent vis-à-vis de ces radiations.
Ils ne prennent pas en compte le maintien de la protection après immersion ou sudation, d’où la nécessité de répéter régulièrement les applications au cours de la journée.
Les indices les plus élevés contre les spectres les plus larges sont à utiliser de préférence en cas de lupus cutané.
Ces photoprotecteurs doivent être systématiquement appliqués tous les jours, même en l’absence d’exposition solaire prévisible dans la journée.
Ils doivent être renouvelés toutes les 2 heures en cas d’exposition solaire.
2- Antipaludéens de synthèse :
En l’absence d’atteinte viscérale de LEAD justifiant un traitement « lourd » (corticothérapie et parfois immunosuppresseurs), le traitement des lupus cutanés fait appel en première intention aux antipaludéens de synthèse, essentiellement à l’hydroxychloroquine et à la chloroquine aux doses de 6,5 mg/kg/j pour l’hydroxychloroquine et de 4 mg/kg/j pour la chloroquine.
L’efficacité n’est pas jugée avant 3 mois de traitement, date à laquelle une amélioration clinique nette est notée dans plus de 80 % des cas.
Leur mode d’action dans les lupus cutanés est mal connu, faisant probablement intervenir un effet photoprotecteur, anti-inflammatoire et immunologique.
Les effets secondaires sont dominés par l’atteinte oculaire.
Les dépôts cornéens sont fréquents et asymptomatiques dans plus de la moitié des cas.
La rétinopathie est plus grave car elle peut retentir sur l’acuité visuelle.
Au stade préclinique, elle se manifeste par une altération de la vision des couleurs avec le plus souvent dyschromatopsie d’axe bleu-jaune.
À un stade plus avancé, existe une maculopathie confirmée avec baisse de l’acuité visuelle en vision paracentrale.
Une fois installée, la maculopathie est irréversible et peut continuer à s’aggraver après l’arrêt du traitement.
L’incidence de cette rétinopathie a diminué depuis plusieurs décennies et est actuellement très faible. Dans la série récente de Lévy, un malade sur 1 207 (0,08 %) avait une toxicité définie et cinq autres (0,4 %) une toxicité probable.
Il existe un consensus pour penser que plusieurs facteurs interviennent dans la prévalence de cette rétinopathie des antipaludéens de synthèse :
– la nature de l’antipaludéen de synthèse, la chloroquine étant plus toxique que l’hydroxychloroquine ;
– les doses quotidiennes prescrites ; les doses considérées à faible risque étant inférieures à 6,5 mg/kg/j pour l’hydroxychloroquine et inférieures à 4 mg/kg pour la chloroquine ;
– la durée du traitement, le risque de toxicité augmentant avec la dose totale ;
– l’âge élevé, l’insuffisance rénale, l’existence d’une atteinte oculaire préalable, éléments considérés comme des facteurs de risques supplémentaires.
Les modalités des examens ophtalmologiques et le rythme de la surveillance oculaire ne font l’objet d’aucun consensus et varient considérablement selon les centres.
En France, l’électrorétinogramme n’est plus considéré comme obligatoire mais reste conseillé par certains ophtalmologistes.
Les autres examens le plus souvent réalisés sont la grille d’Amsler permettant l’autodépistage de pertes du champ visuel, nécessitant une bonne coopération du malade ; la périmétrie statique avec un test automatisé comportant une grille de points couvrant les 10 degrés centraux et espacés de 1 degré, sensible et reproductible ; enfin l’étude de la vision des couleurs avec score chiffré en fonction de l’âge (test D15 désaturé de Lanthony…) autorisant une surveillance dans le temps. Un examen initial est indispensable.
Pendant les 5 premières années de traitement, une surveillance annuelle est souhaitable, alors qu’un examen bisannuel paraît raisonnable au-delà.
Les autres effets secondaires sont plus rares, en dehors de troubles digestifs mineurs (neuromyopathie, agranulocytose, bloc auriculoventriculaire, perte de l’audition…).
Un électrocardiogramme annuel est cependant nécessaire.
La pigmentation cutanée prédominant sur les zones exposées est en revanche relativement fréquente après de nombreuses années de prise d’antipaludéens ; elle peut être également muqueuse, notamment palatine, et unguéale, n’imposant pas l’arrêt du médicament.
Le blanchiment des cheveux est beaucoup plus rare, probablement en rapport avec un effet toxique sur le mélanocyte.
Un prurit, une urticaire, une vascularite ou un exanthème maculopapuleux ont été signalés dans des cas isolés.
La contreindication des antipaludéens pendant la grossesse n’est plus reconnue aujourd’hui par une majorité d’auteurs.
En cas d’échec apparent d’un traitement par antipaludéens de synthèse, il faut s’assurer de la prise correcte du médicament, de l’absence de facteurs inducteurs tels que des expositions solaires et combattre le tabagisme, considéré comme un facteur de résistance aux antipaludéens.
L’augmentation des doses d’antipaludéens augmente l’efficacité mais aussi le risque de toxicité ; des doses élevées ne doivent pas être prescrites sur de longues durées.
Le changement de l’hydroxychloroquine par la chloroquine ou l’inverse peut permettre de contrôler les lésions dans un pourcentage de cas qui reste à déterminer.
Aux États-Unis, l’association de plusieurs antipaludéens de synthèse entre eux, telle que l’association chloroquine-quinacrine ou hydroxychloroquine-quinacrine est prescrite en deuxième intention. Environ 100 à 200 mg de quinacrine sont alors ajoutés au traitement antérieur.
La quinacrine a des effets secondaires voisins de ceux des deux autres antipaludéens, mais sans toxicité oculaire.
Elle donne une coloration jaune pigmentée des téguments, parfois considérée comme inesthétique.
L’apparition d’une réaction lichénoïde doit faire arrêter le traitement car elle peut précéder une toxicité médullaire.
L’efficacité de cette association n’a été rapportée que dans des études ouvertes limitées.
Paradoxalement a été notée une toxicité oculaire majorée.
L’absence de commercialisation de la quinacrine limite son utilisation en France.
3- Corticothérapie :
La corticothérapie locale (dermocorticoïde de niveau I) est réservée aux formes limitées.
La corticothérapie générale n’est pas indiquée ; son activité est en effet médiocre sur les lésions cutanées avec une corticodépendance très fréquente.
4- Thalidomide :
En France, le médicament utilisé en deuxième intention est le thalidomide à la dose initiale de 100 mg/j.
Son efficacité n’a été évaluée que dans des études ouvertes avec une rémission des lésions dans plus de 70 % des cas obtenue en moins de 3 mois.
Cette rémission est transitoire avec des rechutes dans presque tous les cas à l’arrêt du thalidomide.
Aussi est-il nécessaire de prescrire une dose d’entretien la plus faible possible.
Les règles de prescription du thalidomide sont très strictes (médecin dûment autorisé, distribution hospitalière exclusive, pratique systématique d’un test de grossesse tous les 2 mois et contraception efficace obligatoire chez la femme en période d’activité ovarienne, procréation interdite chez l’homme, lecture et signature d’un document informant sur les risques tératogènes encourus, normalité de l’électromyogramme avec étude des vitesses de conduction nerveuse).
Le thalidomide peut induire une somnolence mieux acceptée en cas de prise le soir, une prise de poids, une aménorrhée ou une impuissance chez l’homme.
Les risques de neuropathie axonale sensitive et distale sont non négligeables, contre-indiquant ce traitement chez certains sujets prédisposés (alcooliques, diabétiques…).
Une surveillance neurologique clinique mensuelle et électromyographique bisannuelle est préconisée.
5- Dapsone :
La dapsone à la dose de 100 à 150 mg/j a permis de blanchir certains malades atteints de lupus discoïde ou de LES.
De faibles doses (inférieures à 100 mg/j) sont souvent suffisantes dans les LES, ce qui permet de diminuer la toxicité, en particulier l’hémolyse et la méthémoglobinémie dose-dépendantes.
Rappelons qu’il est nécessaire, avant de prescrire ce médicament, de vérifier l’absence de déficit en G6PD, surtout dans certaines ethnies, et la possibilité au cours du traitement de survenue d’un syndrome d’hypersensibilité, heureusement rare.
La prescription concomitante de foldine en améliore la tolérance.
6- Rétinoïdes :
Les rétinoïdes, en particulier l’acitrétine (Soriatanet) ou l’isotrétinoïne (Roaccutanet), à la dose de 0,5 à 1 mg/kg/j, sont une alternative thérapeutique pour les lupus cutanés résistants.
Leur efficacité n’est pas limitée au lupus verruqueux ; elle a été également rapportée chez des malades ayant un lupus discoïde ou un LES dans des séries ouvertes de taille limitée.
La longue durée (2 ans) de la contraception imposée par la prise d’acitrétine conduit à choisir plutôt chez une jeune femme l’isotrétinoïne. Les effets secondaires des rétinoïdes en limitent l’utilisation.
7- Clofazimine :
La clofazimine (Lamprènet) à la dose de 100 mg/j a été employée avec succès dans des cas isolés de lupus discoïde ou de LES.
Il peut induire une coloration brune inesthétique, notamment des lésions séquellaires atrophiques de lupus discoïde.
8- Autres :
L’IFN-alpha a été utilisé à une dose de 18 à 120 millions d’unités par semaine pendant 4 à 13 semaines chez des malades atteints de lupus discoïde ou de LES.
L’amélioration des lésions cutanées, constatée dans plus de 50 % des cas, a été transitoire, associée à une stimulation de l’auto-immunité dangereuse sur ce terrain. Rappelons que l’IFN-alpha peut induire des lupus.
Les multiples effets secondaires des sels d’or, en particulier cutanéomuqueux (réaction lichénoïde, érythrodermie exfoliative…) ont fait pratiquement abandonner sa prescription dans les lupus cutanés, alors que des améliorations avaient été rapportées antérieurement.
La salazopyrine à la dose de 1,5 ou 2 g/j a donné des résultats très satisfaisants dans près de la moitié des cas de séries ouvertes de malades atteints de lupus discoïde.
Les effets secondaires sont cependant nombreux, parfois graves à type de syndrome d’hypersensibilité ou d’exacerbation du lupus.
L’efficacité thérapeutique serait associée au phénotype d’acétylation rapide qui devrait être systématiquement recherché avant de débuter le traitement.
L’efficacité de la phénytoïne n’a été étudiée que dans une seule étude de 93 malades atteints de lupus discoïde, non sélectionnés du fait de leur résistance.
Après 4 mois de traitement, seulement 50 malades sont analysables avec obtention d’une rémission totale dans 88 % des cas et des effets secondaires mineurs.
En raison des effets secondaires connus de la phénytoïne, d’autres études concordantes sont indispensables avant d’envisager de prescrire ce médicament chez des malades atteints de lupus cutané résistant.
Les immunosuppresseurs ont été prescrits dans des cas anecdotiques avec un bon effet, notamment l’azathioprine et le méthotrexate.
Ils ne sont envisagés qu’en dernier recours chez des malades avec un retentissement psychologique ou une impotence fonctionnelle majeure.
Il en est de même des gammaglobulines qui, dans notre expérience, ont eu un effet favorable mais souvent suspensif chez huit malades sur neuf souffrant d’un lupus discoïde et chez un malade avec
LES particulièrement résistant. Enfin, quelques malades avec LES ont été améliorés par une irradiation généralisée à faibles doses d’UVA-1 (340-400 nm).
Étant donné le risque théorique élevé d’aggravation du LES par cette irradiation, l’indication de cette UVA-1-thérapie reste encore très controversée.
Des études contrôlées appréciant les effets bénéfiques et délétères de ces produits sont indispensables pour guider le choix thérapeutique des lupus cutanés résistant aux antipaludéens de synthèse.
Lésions vasculaires :
Elles sont principalement observées dans les LEAD.
En dehors des acrosyndromes et des oedèmes angioneurotiques, elles sont secondaires à une atteinte inflammatoire (vasculite) ou thrombotique des vaisseaux cutanés.
Un diagnostic précis est indispensable étant donné les conséquences thérapeutiques totalement opposées.
La mise en évidence d’une thrombose impose la recherche d’anticorps antiphospholipides.
A – ACROSYNDROMES :
Un phénomène de Raynaud est présent chez 10 à 45 % des malades atteints de LEAD, pouvant précéder de longue date l’apparition des autres symptômes.
Il serait encore plus fréquent (60 à 90 %) chez les malades avec anticorps anti-U1 RNP. Il ne justifie que rarement un traitement spécifique.
L’apparition d’une nécrose digitale doit faire suspecter une thrombose ou une vasculite associée. D’exceptionnels cas d’érythermalgies ont été rapportés au cours de LEAD, avec une bonne efficacité du clonazépam.
B – LIVEDO :
Autrefois considéré comme une manifestation de vasculite lupique, le livedo est en fait statistiquement associé au cours du lupus à la présence d’anticorps antiphospholipides et aux manifestations vasculaires ischémiques cérébrales.
Ce livedo est habituellement diffus, non infiltré, à mailles fines non fermées formant des cercles incomplets (livedo racemosa ou livedo ramifié), localisé sur les membres et surtout le tronc.
Les biopsies cutanées sur les mailles ou entre les mailles sont le plus souvent normales ; ailleurs elles mettent en évidence une artériolopathie oblitérante non spécifique, exceptionnellement une thrombose.
C – ULCÈRES DE JAMBES :
Ils sont observés chez 3 % environ des malades ayant un LEAD.
Ils imposent de pratiquer un doppler artériel et veineux des membres inférieurs, ainsi qu’une biopsie des bords pour en comprendre le mécanisme, vasculite ou plus souvent thrombose profonde ou superficielle.
Leur fréquence est en effet incontestablement plus élevée en présence d’anticorps antiphospholipides, allant de 5 à 39%.
D – URTICAIRE ET OEDÈME DE QUINCKE :
Des lésions d’urticaire ont été notées dans 4 à 13 % des grandes séries de LEAD, correspondant histologiquement à une vasculite leucocytoclasique des vaisseaux superficiels dermiques.
Ces lésions urticariennes sont souvent associées à un complément abaissé et à des anticorps anti-C1q, par ailleurs très fréquemment observés au cours du LEAD.
Elles peuvent s’accompagner de lésions d’oedème de Quincke, à différencier alors de l’oedème angioneurotique, en rapport avec un déficit congénital de l’inhibiteur de la C1-estérase dont la prévalence est augmentée dans le LEAD.
E – HÉMORRAGIES EN FLAMMÈCHES MULTIPLES SOUS-UNGUÉALES :
Leur survenue brutale sur plusieurs doigts au cours d’un LEAD témoigne le plus souvent d’un événement important systémique tel qu’une thrombose profonde ou une poussée lupique.
Leur mécanisme reste encore hypothétique : embolie, vasculite ou thrombose ?
F – NÉCROSES CUTANÉES EXTENSIVES :
Leur début est volontiers brutal avec un purpura nécrotique laissant rapidement place à une plaque escharrotique noirâtre bordée d’un liseré purpurique témoignant de leur évolutivité.
Elles sont localisées sur les membres, le visage (joues, nez, oreilles) ou les fesses.
La biopsie de la bordure purpurique objective aisément des thromboses multiples.
Le traitement fait appel à l’anticoagulation, aux vasodilatateurs dérivés de la prostacycline et éventuellement aux échanges plasmatiques.
G – AUTRES LÉSIONS VASCULAIRES :
D’autres lésions vasculaires peuvent survenir au cours d’un LEAD.
Certaines sont de mécanisme incertain car non biopsiées.
Il en est ainsi de l’érythème palmaire, lunulaire et des télangiectasies périunguéales ressemblant à celles observées au cours des dermatomyosites souvent accompagnées de mégacapillaires à la capillaroscopie, observés chez 10 à 15 % des malades avec LEAD.
Les lésions purpuriques infiltrées plus ou moins nécrotiques peuvent correspondre à une vasculite ou à des thromboses.
Quant aux lésions atrophiques ivoirines dites d’atrophie blanche ou de pseudomaladie de Degos, elles semblent plus souvent d’origine thrombotique que vasculitique, observées essentiellement en présence d’anticorps antiphospholipides.
Manifestations non lupiques, non vasculaires :
Elles forment un groupe hétéroclite de manifestations dermatologiques préférentiellement observées au cours des lupus.
Certaines sont fréquentes, telle l’alopécie, alors que d’autres sont rares comme le lupus bulleux, la mucinose ou la pustulose amicrobienne.
A – LUCITE IDIOPATHIQUE :
Les lucites idiopathiques telles que la lucite estivale bénigne ou la lucite polymorphe sont très fréquentes dans la population générale, atteignant près de 20 % de la population scandinave.
La plupart des études épidémiologiques ont démontré que la présence d’une telle lucite n’augmentait pas le risque de développer ultérieurement un lupus.
Cependant, deux études de la même équipe finlandaise ont mis en évidence une histoire de lucite idiopathique ou des phototests compatibles avec ce type de réaction chez la moitié des malades avec diverses formes de lupus cutané.
La lucite idiopathique précédait dans la moitié des cas les lésions lupiques. Inversement, la prévalence des lupus chez les malades avec lucite idiopathique paraît faible, estimée à 2 %.
Dans notre expérience, les lucites idiopathiques ne semblent pas plus fréquentes chez les lupiques que dans la population générale.
En revanche, il est parfois difficile de différencier une lucite polymorphe de lésions lupiques.
B – ALOPÉCIE :
Dans le LEAD, il ne s’agit pas d’une alopécie cicatricielle secondaire à des lésions lupiques mais d’une chute diffuse des cheveux (effluvium télogène) contemporaine des poussées ou survenant 3 mois après, pouvant donner un cuir chevelu clairsemé, disparaissant progressivement après traitement. Ailleurs les cheveux sont fins et fragiles, facilement cassés.
Il peut exister alors une bande de cheveux de ½ cm de longueur (cheveux lupiques) en bordure du cuir chevelu (front, tempes).
C – LUPUS BULLEUX :
Il se manifeste cliniquement par des bulles ou des vésiculobulles, parfois regroupées en bouquets, apparaissant en peau saine sur les zones exposées et non exposées, disparaissant sans cicatrice ni grain de milium.
Histologiquement, il s’agit de bulles sousépidermiques avec un infiltrat de polynucléaires neutrophiles et éosinophiles et souvent une vascularite leucocytoclasique dermique.
L’immunofluorescence directe est généralement positive avec des dépôts d’IgG ou d’IgM et d’IgA à la jonction dermoépidermique.
Le clivage de la bulle est dermique superficiel en microscopie électronique. Biologiquement existent des anticorps anticollagène de type VII (protéine majeure de 290 kDa et mineure de 145 kDa en western blot).
Les lésions bulleuses disparaissent habituellement avec la dapsone.
Le lupus bulleux est à différencier des bulles par nécrose épidermique au cours du LEA ou du LEAD et des rares associations de LEAD avec d’autres maladies bulleuses auto-immunes : pemphigoïde bulleuse, pemphigus, dermatite herpétiforme, dermatose bulleuse à IgA linéaire…
D – MUCINOSE PAPULEUSE :
Alors que des dépôts de glycosaminoglycanes sont fréquemment mis en évidence par l’histologie au sein des lésions cutanées lupiques comme dans les dermatomyosites, la présence isolée de tels dépôts dans le derme sans lésion lupique est plus rare.
Elle se manifeste par des lésions papuleuses, plus rarement nodulaires, localisées préférentiellement sur le cou, la racine des membres supérieurs et le tronc.
Cette mucinose papuleuse serait présente dans certaines séries chez 1,5 % des malades lupiques.
Elle est observée plus fréquemment dans le LEAD (65 %) que dans les LEC sans manifestation systémique (35 %).
Les dépôts de glycosaminoglycanes sont localisés dans le derme superficiel et moyen, entourés d’un discret infiltrat lymphocytaire.
Les antimalariques ne seraient efficaces que dans 20 % des cas.
L’abstention thérapeutique ou l’emploi de corticostéroïdes peuvent être discutés.
E – ANÉTODERMIE :
Les lésions d’anétodermie sont définies histologiquement par la disparition localisée du tissu élastique, non centrée par un follicule pileux, sur toute la hauteur du derme, et cliniquement par l’existence d’un phénomène de herniation à la palpation.
Le nombre et la taille des lésions sont excessivement variables.
Elles sont surtout localisées sur le cou et la moitié supérieure du tronc et des bras.
Au sein du lupus a été récemment soulignée l’association préférentielle de ces lésions avec la présence d’anticorps antiphospholipides et la possibilité de microthromboses en histologie.
F – CALCIFICATIONS :
Les calcifications cutanées sont beaucoup plus rares dans le lupus que dans la sclérodermie.
Leur présence doit faire rechercher une connectivite mixte et la présence d’anticorps anti-U1RNP.
Elles peuvent être observées en regard des lésions lupiques ou à distance.
G – PUSTULOSE AMICROBIENNE DES PLIS :
Une pustulose amicrobienne des grands et petits plis associée à des pustules isolées du cuir chevelu a été récemment décrite au cours du lupus et d’autres maladies auto-immunes.
L’aspect histologique est celui d’une pustule spongiforme.
Les surinfections sont fréquentes, avec un aspect suintant, notamment de la région génitale.
Un déficit en zinc a été rapporté dans quelques cas.
Les lésions sont sensibles à la corticothérapie générale ou locale.
Conclusion :
L’analyse sémiologique rigoureuse des lésions dermatologiques observées au cours des lupus, au besoin complétée par un examen anatomopathologique d’un prélèvement biopsique, doit permettre un diagnostic précis indispensable avant de proposer un traitement adapté.







")



{kind=link}