



















I – DÉTERMINATION DU STADE ÉVOLUTIF :
1- Classification TNM des lymphomes cutanés T épidermotropes :
La classification TNM est, de loin, la plus utilisée dans les lymphomes cutanés épidermotropes.
2- Stade des lymphomes cutanés T épidermotropes :
Cette détermination en quatre stades a été proposée bien avant la nouvelle classification EORTC des lymphomes cutanés.
Elle intéresse l’ensemble des lymphomes cutanés épidermotropes et s’applique donc aussi au syndrome de Sézary.
J – ÉVOLUTION ET PRONOSTIC :
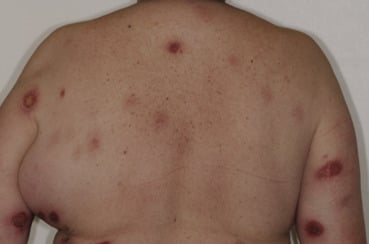 L’évolution s’étend sur des années, voire des décennies.
L’évolution s’étend sur des années, voire des décennies.
Le décès est le plus souvent la conséquence d’une surinfection microbienne ou virale.
Il peut également résulter de l’envahissement tumoral et/ou de la transformation en un lymphome à grandes cellules.
Le taux de survenue d’une transformation en lymphome à grandes cellules varie, selon les séries, de 8 % à plus de 55 %.
Cette transformation, possible à n’importe quel stade de la maladie, est d’autant plus fréquente que le stade de la maladie est avancé.
Elle est ainsi beaucoup plus fréquente aux stades IIb et IV qu’aux stades I et IIa.
Son pronostic est d’autant plus sombre qu’elle est survenue à un stade précoce de la maladie.
Le taux de survie à 5 ans de 278 patients inclus dans le groupe néerlandais d’étude des lymphomes cutanés est de 87 %, tous stades confondus.
Un cancer secondaire est observé dans 5,7 % des cas.
Les facteurs pronostiques les plus fiables sont représentés par le T de la classification TNM, et par l’épaisseur de la lésion cliniquement la plus infiltrée.
La différence entre les formes cutanées précoces (T1, T2) et les formes avancées (T4) apparaît clairement dans la littérature.
Les malades au stade des plaques sans atteinte ganglionnaire, sanguine ou viscérale ont une moyenne de survie supérieure à 5 ans.
Ceux avec localisation ganglionnaire ou sanguine, sans atteinte viscérale et sans effacement de l’architecture ganglionnaire, ont une moyenne de survie de 5 ans.
Enfin, ceux avec envahissement viscéral ou effacement de l’architecture ganglionnaire ont une moyenne de survie de 2 ans et demi. Les études génotypiques auraient également une valeur pronostique.
Ainsi, parmi les patients porteurs d’adénomégalies, elles permettraient de distinguer ceux pour lesquels le pronostic est mauvais car une population clonale est détectée dans leurs ganglions de ceux dont le pronostic est meilleur car l’étude génotypique ganglionnaire est normale.
De la même manière, la détection de populations monoclonales T dans des lésions cutanées évocatrices de mycosis fongoïde débutant indiquerait un risque évolutif plus grand.
Toutefois, ces résultats n’ont pas de conséquence pratique pour le moment.
En effet, en l’absence d’études prospectives, on ne sait pas si le traitement précoce et incisif des formes dans lesquelles une clonalité est détectée améliore ou non le devenir des patients.
K – TRAITEMENT :
La rareté du mycosis fongoïde, sa longue durée d’évolution spontanée qui s’étend sur des années, voire des décennies, l’existence de variantes cliniques de la maladie enfin rendent difficiles les études comparatives de ses différents traitements.
Les études thérapeutiques utilisant des groupes contrôles sont très difficiles à mettre en oeuvre et ne peuvent se concevoir que de manière multicentrique, à l’échelon international.
Une revue de la littérature concernant le traitement du mycosis fongoïde a été réalisée en 1997 par Richard-Lallemand et Carsuzaa.
Près de 200 articles y sont référencés.
1- Moyens thérapeutiques :
* Moyens locaux :
+ Corticothérapie locale :
Aux stades de début du mycosis fongoïde, au stade IA en particulier, on peut utiliser les dermocorticoïdes en application biquotidienne.
Il est recommandé d’utiliser des dermocorticoïdes d’activité très forte et d’avoir éventuellement recours aux pansements occlusifs.
+ PUVA-thérapie :
Elle associe une prise orale de comprimés de 8-méthoxy-psoralène à la dose de 0,6 mg/kg et, 2 heures plus tard, une irradiation UVA.
La dose initiale d’UVA est de 1,5 à 3 J/cm2 selon le phototype, augmentée de 0,5 J/cm2 à chaque séance en l’absence d’érythème.
Elle est réalisée au rythme de trois séances par semaine au début. Une fois les lésions cutanées disparues, cliniquement et histologiquement, un traitement d’entretien avec des séances régulièrement espacées est nécessaire.
Les principaux effets secondaires sont la survenue de prurit, de nausées, d’érythème avec sécheresse cutanée et la réactivation possible d’infection à Herpès virus.
Cette méthode a l’avantage d’être bien acceptée des malades sur le plan pratique.
Malheureusement, elle expose à la survenue de carcinomes spinocellulaires.
+ Chimiothérapie locale :
La méchloréthamine (Caryolysinet) est utilisée soit en solution aqueuse, soit sous forme d’une base anhydre.
On utilise, en général, une concentration de 0,02 %.
Les applications doivent être faites dans une pièce aérée.
Il faut porter des gants en PVC, éviter le contact direct avec la méchloréthamine.
Si le patient procède luimême aux applications, il ne doit pas oublier le haut du dos, le dos des mains et les faces antérieures des deux jambes.
Cette méthode a pour avantage de ne nécessiter aucun équipement spécial. Une réaction d’hypersensibilité retardée survient dans près de 40 % des cas en moins de 1 an de traitement.
Les essais d’induction de tolérance se sont révélés inefficaces ; les désensibilisations topiques sont en général efficaces.
Le risque de sensibilisation de contact est moindre avec la base anhydre qu’avec la solution aqueuse.
Les réactions urticariennes sont plus rares. Elles doivent conduire à l’arrêt du traitement par crainte de survenue d’une réaction générale de type anaphylactique.
Au total, ce traitement est simple, peu coûteux et efficace.
Il peut induire la survenue, à long terme, de tumeurs cutanées (carcinome baso- et spinocellulaire, nævi dysplasiques).
La carmustine (Bicnut) topique est une alternative à la méchloréthamine.
Préparé en solution à 0,2 % dans l’alcool absolu, dilué deux fois au moment de l’emploi, ou en onguent anhydre à 0,4 %, il s’applique tous les jours en début de traitement.
Les effets secondaires habituels sont l’érythème, l’impression de sécheresse cutanée et les télangiectasies.
Sa myélotoxicité éventuelle témoigne de son absorption percutanée
+ Électronthérapie superficielle :
La technique d’électronthérapie superficielle totale a avantageusement remplacé les techniques d’irradiation corporelle totale par radiothérapie superficielle.
Elle est utilisée soit isolément, soit en combinaison avec d’autres traitements.
Elle peut être répétée.
Les effets secondaires, importants mais transitoires, sont à type de sécheresse, de desquamation, de fissuration cutanée, d’oedèmes, voire de décollements bulleux.
On peut également observer une chute des ongles et des poils, un état poïkilodermique, voire des radiodermites.
Le risque carcinogène à long terme est élevé puisque Licata et al ont dénombré 6 % de mélanomes et 21 % de carcinomes.
Le risque de carcinomes est encore augmenté par l’utilisation, auparavant ou en même temps, d’autres traitements locaux tels que PUVA-thérapie ou badigeons de Caryolysinet.
L’électronthérapie superficielle localisée a pour indication principale le mycosis fongoïde unilésionnel, pour lequel l’électronthérapie superficielle totale n’est pas indiquée.
* Moyens généraux :
+ Chimiothérapies :
De très nombreuses chimiothérapies ont été utilisées dans le traitement des lymphomes cutanés.
Les molécules les plus utilisées en monothérapie sont le méthotrexate à faibles doses, le cyclophosphamide, et le chlorambucil associé ou non à la prednisone.
Les analogues des purines utilisées sont la 2’déoxycoformicine (pentostatine), inhibiteur de l’adénosine désaminase, relativement spécifique des lymphocytes T et la 2’chlorodéoxyadénosine (fludarabine).
Les molécules les plus utilisées en polychimiothérapie sont le cyclophosphamide, la doxorubicine, la vincristine, la bléomycine et le méthotrexate.
De très nombreux protocoles de polychimiothérapies intermittentes cycliques ont été utilisés : CVBP, CHOP/HOP, COP, CAVOP, CVP et CBP.
+ Rétinoïdes :
Trois dérivés différents de la vitamine A, dotés d’une activité à la fois antitumorale et immunomodulatrice, ont été utilisés : l’étrétinate (Tigasont), l’isotrétinoïne (Roaccutanet) et l’arotinoïde ester (Atrat) à des posologies variant entre 0,5 et 1,5 mg/kg.
Les effets secondaires, communs à tous ces rétinoïdes (sécheresse cutanéomuqueuse, syndrome dépressif, élévation des transaminases, du cholestérol et des triglycérides), sont dose-dépendants.
+ Interféron-alpha :
Doté d’une activité antiproliférative et immunomodulatrice, il est administré en injections sous-cutanées ou intratumorales, à raison de trois fois par semaine, à des doses de 3 à 12 millions d’unités par injection.
Les effets secondaires, d’autant plus à craindre que les patients sont âgés, sont dose dépendants (fièvre, syndrome grippal, asthénie, anorexie, troubles neuropsychiques avec dépression, élévation des transaminases, du cholestérol et des triglycérides, leucopénie et thrombopénie).
2- Indications thérapeutiques :
Il n’existe pas de schéma thérapeutique précisément établi.
Les indications thérapeutiques doivent être envisagées essentiellement en fonction du stade évolutif du mycosis fongoïde.
* Stades I et II :
À ces stades de début, on a recours aux thérapeutiques « externes », c’est-à-dire à impact cutané préférentiel, telles que la photochimiothérapie, les applications locales d’antimitotiques ou l’électronthérapie. Des rétinoïdes peuvent être associés à la photochimiothérapie.
L’interféron peut également être utilisé.
+ Dermocorticoïdes :
Ils donnent plus de 90 % de réponses globales, dont près de deux tiers de réponses complètes au stade T1, et encore 80 % de réponses globales dont un quart de rémissions complètes au stade T2.
+ Photothérapie :
Avec la PUVA-thérapie, une rémission complète est obtenue dans environ 70 % des cas au stade I et 50 % au stade II.
Des rémissions de longue durée sont possibles.
Tous les auteurs insistent sur la nécessité d’un traitement d’entretien.
L’UVB-thérapie est surtout efficace au stade des érythèmes non infiltrés.
L’association rétinoïdes-PUVAthérapie permet d’obtenir des rémissions complètes dans un pourcentage de cas équivalent mais avec une dose d’UVA moindre, le taux de rechutes après rémission complète étant équivalent.
+ Chimiothérapie locale :
Avec les badigeons de méchloréthamine (Caryolysinet), une rémission complète est obtenue chez 75 % des malades avec lésions non infiltrées et chez 50 % de ceux avec lésions infiltrées.
Le taux et la durée des rémissions complètes sont comparables à ceux obtenus avec la PUVA-thérapie.
En cas de plaques limitées, bains d’électrons et méchloréthamine donnent des résultats identiques.
La durée des rémissions apparaît plus longue qu’avec les bains d’électrons.
Le taux de 10 à 30 % de rémissions à 8 ans ne peut être considéré comme un pourcentage de guérisons en l’absence d’étude prospective au long cours chez des malades traités uniquement et d’emblée par la moutarde à l’azote.
Avec la carmustine (Bicnut), le délai de réponse est plus court qu’avec la Caryolysinet, avec des taux de rémissions complètes identiques et des rémissions plus longues.
+ Électronthérapie :
Avec l’électronthérapie corporelle totale, une rémission complète est obtenue chez 90 % des patients au stade I et chez 70 % au stade IIA.
L’élément le plus significatif du pronostic est représenté par la présence ou l’absence d’adénopathies palpables.
Pour certains auteurs, de plus hautes doses d’irradiation n’améliorent pas le taux de survie ; d’autres considèrent que si, à condition que les lésions cutanées soient localisées et d’extension limitée et qu’il n’y ait pas d’envahissement ganglionnaire.
L’association électronthérapie corporelle totale et irradiation ganglionnaire augmente la durée des rémissions complètes mais pas le taux de survie.
Aucune étude ne permet d’affirmer que ce traitement puisse être curatif, même si des rémissions prolongées sont observées. Plusieurs associations thérapeutiques ont été essayées.
L’association avec des rétinoïdes n’augmente pas le taux des rémissions complètes.
L’adjonction d’une polychimiothérapie, une fois la rémission complète obtenue, n’en augmente pas la durée.
Enfin, si l’association électronthérapie superficielle totale et polychimiothérapie permet d’obtenir un taux de rémissions complètes supérieur à celui résultant d’un simple traitement par badigeons de Caryolysinet, il n’y a pas de différence significative tant en termes de durée de rémissions qu’en termes de survie.
+ Chimiothérapie générale :
Un très petit nombre de patients à des stades précoces (IA, IIA) ont été traités en première intention par une chimiothérapie générale et, si quelques patients ont des rémissions complètes de longue durée, le recul n’est pas suffisant pour savoir s’il s’agit de rémissions prolongées ou de véritables guérisons.
+ Rétinoïdes et interféron-alpha :
Aux stades précoces du mycosis fongoïde, les rétinoïdes donnent 60 % de réponses mais ces réponses sont de courte durée.
Les interférons-alpha donnent 20 % de rémissions complètes et 40 % de rémissions partielles.
Certaines semblent durables. L’association rétinoïdes/interféron donne 50 % de réponses.
Avec l’association interféron-alpha/PUVA-thérapie, on obtient 90 % de réponses, dont plus de 60 % de rémissions complètes.
Le taux de rémissions complètes chez les patients traités en première intention par l’association interféron-alpha/PUVA-thérapie semble supérieur à celui obtenu avec l’interféron seul.
* Stade III :
Le mycosis érythrodermique bénéficie de la même stratégie thérapeutique que le syndrome de Sézary.
* Stade IV :
Les taux de réponse sont importants mais de courte durée, la durée étant plus longue avec une polychimiothérapie qu’avec une monochimiothérapie.
La médiane de survie, de 6 mois avec une polychimiothérapie, n’est pas supérieure à celle des malades non traités.
Variantes de mycosis fongoïde :
À côté de la forme classique de mycosis fongoïde, il existe de nombreuses variantes cliniques, comme le mycosis fongoïde bulleux ou le mycosis fongoïde hyper- et hypopigmenté, qui ne sont pas considérées à part dans la classification EORTC 1997, car elles ont un comportement clinique similaire.
En revanche, le mycosis fongoïde associé à une mucinose folliculaire, le mycosis fongoïde avec tropisme particulier pour les annexes cutanées, follicules pileux ou glandes sudorales (formes dites respectivement pilotropes et syringotropes), le lymphome pagétoïde et la chalazodermie granulomateuse, entités appartenant au groupe des lymphomes cutanés dits « épithéliotropes », ont des caractéristiques clinicopathologiques distinctes et sont, par là même, considérés à part dans la classification EORTC 1997.
A – MYCOSIS FONGOÏDE ASSOCIÉ À UNE MUCINOSE FOLLICULAIRE ET LYMPHOMES PILOTROPES :
1- Définition :
Cette variante est caractérisée par la présence d’un infiltrat folliculotrope épargnant l’épiderme, avec ou sans dégénérescence mucineuse des follicules pileux, et atteignant préférentiellement l’extrémité cervicocéphalique.
2- Clinique :
Des papules folliculaires, des plaques indurées et des tumeurs sont souvent associées avec une chute des poils et avec un prurit.
Les surinfections bactériennes sont fréquentes.
Dans les lymphomes pilotropes, on observe également des lésions pseudocomédoniennes et des kystes épidermiques.
Ces lésions folliculaires peuvent révéler le lymphome, survenir en même temps que des lésions de mycosis fongoïde classique, voire précéder ou accompagner un épisode de rechute de mycosis fongoïde.
3- Histopathologie :
L’infiltrat dermique a une localisation essentiellement périvasculaire et périannexielle avec une infiltration marquée de l’épithélium folliculaire par les cellules de moyenne à grande taille à noyau hyperchromatique et cérébriforme.
Des dépôts de mucine, colorés en bleu par le bleu Alcian, sont observés dans l’épithélium folliculaire envahi.
On peut observer, au sein de l’infiltrat, la présence d’éosinophiles et d’autres cellules inflammatoires.
Des cas similaires, sans mucinose folliculaire, ont été rapportés comme mycosis fongoïdes folliculaires ou comme mycosis fongoïdes pilotropes.
Lorsque l’épiderme est épargné, on parle de pilotropisme ou de folliculotropisme au lieu d’épidermotropisme.
En l’absence d’épidermotropisme, ces formes de mycosis fongoïde, associées ou non à une mucinose folliculaire, peuvent être méconnues et classées incorrectement comme lymphomes T pléomorphes.
Quand des lésions pseudocomédoniennes ou des kystes épidermiques sont présents, l’infiltrat et le pilotropisme diminuent.
Aussi, des biopsies multiples peuvent-elles être nécessaires pour arriver au diagnostic.
4- Immunophénotype :
Il est similaire à celui du mycosis fongoïde. ¦ Génotypie Des réarrangements clonaux du gène du TCR sont mis en évidence dans la plupart des cas.
5- Évolution et pronostic :
Des lésions de mucinose folliculaire (alopécie mucineuse) peuvent s’accompagner secondairement d’un lymphome dans au moins 15 % des cas ; il s’agit souvent d’un mycosis fongoïde, parfois d’une maladie de Hodgkin.
Il n’y a pas, à l’heure actuelle, de critères histologiques, cliniques et génotypiques permettant de distinguer la forme primitive isolée de mucinose folliculaire et la forme secondaire associée aux lymphomes.
Le taux de survie à 5 ans de 30 patients inclus dans le groupe néerlandais d’étude des lymphomes cutanés est de 70 %.
6- Traitement :
Parce que l’infiltrat dermique est de localisation périfolliculaire, le mycosis fongoïde associé à une mucinose folliculaire est souvent moins sensible aux thérapeutiques à impact cutané (PUVA-thérapie) que le mycosis fongoïde classique en plaques.
Les résultats avec la méchloréthamine sont discordants selon les équipes.
L’électronthérapie corporelle totale et l’association rétinoïdeinterféron semblent être les meilleurs traitements.
B – LYMPHOME PAGÉTOÏDE :
1- Définition – Historique :
Autre variante clinique rare de mycosis fongoïde, le lymphome pagétoïde, encore appelé réticulose pagétoïde, se caractérise cliniquement par des lésions circinées, bien limitées et, histologiquement, par une prolifération essentiellement localisée dans l’épiderme.
Deux types de lymphome pagétoïde ont été décrits : un type localisé (forme de Woringer-Kolopp) en 1939 et un type disséminé avec lésions multiples et localisations ganglionnaires et viscérales (forme de Ketron-Goodman) en 1931, lequel se comporte comme un mycosis fongoïde classique.
Ce dernier type ayant la même évolutivité qu’un mycosis fongoïde classique, le terme de lymphome pagétoïde ne doit s’appliquer qu’au type localisé de Woringer-Kolopp.
2- Clinique :
Il s’agit d’une lésion unique (forme de Woringer-Kolopp) en plaque érythématosquameuse, hyperkératosique, voire verruqueuse, siégeant le plus souvent sur l’extrémité d’un membre et d’évolution prolongée, lentement progressive.
Il n’a jamais été rapporté de localisation extracutanée dans cette forme.
3- Histopathologie :
L’épidermotropisme de l’infiltrat est massif, avec une acanthose et une désagrégation des couches basales de l’épiderme.
Les cellules de l’infiltrat sont de taille moyenne à grande (15 à 20 ím). Dans l’épiderme, elles sont isolées ou disposées en nids ou en amas, d’où le terme de « pagétoïde ».
Leur noyau est quelquefois hyperchromatique et convoluté, et leur cytoplasme est abondant et vacuolisé.
L’infiltrat dermique est plus polymorphe que celui observé dans le mycosis fongoïde.
4- Immunophénotype :
Les cellules tumorales expriment le phénotype CD3+, CD4+, CD8– ou, dans 50 % des cas, le phénotype CD3+, CD4–, CD8+, voire CD4–, CD8–.
L’antigène CD30 peut être exprimé. Dans quelques cas, une perte de l’expression de l’antigène CD7 a été rapportée.
5- Génotype :
Un réarrangement clonal du gène du TCR est habituellement détecté.
6- Évolution – Pronostic :
Le pronostic est excellent. Il n’a jamais été rapporté de localisations extracutanées.
Des récurrences locales après traitement ont toutefois été signalées.
Le décès en rapport avec la maladie n’ayant jamais été rapporté, le concept de « lymphome cutané bénin » a été introduit.
7- Traitement :
Il consiste en l’excision chirurgicale, avec une marge d’exérèse de 0,5 cm, suivie d’une radiothérapie externe localisée et éventuellement d’une PUVA-thérapie.
Syndrome de Sézary :
A – DÉFINITION :
Sézary a décrit, en 1938, un syndrome particulier associant, à la phase d’état, une érythrodermie sèche ou oedémateuse à gros plis cutanés, souvent pigmentée, très prurigineuse, une polyadénopathie superficielle et la présence dans le sang de cellules mononucléées monstrueuses qui portent son nom.
Il n’existe pas de consensus concernant cette définition.
Certains auteurs n’hésitent pas à assimiler tous les lymphomes à cellules T érythrodermiques au syndrome de Sézary, y compris le mycosis fongoïde érythrodermique. Nous définissons le syndrome de Sézary comme l’association à une érythrodermie clinique d’un contingent monoclonal de cellules de Sézary circulantes.
B – CLINIQUE :
Aux signes précédemment cités dans la définition peuvent s’ajouter une onychodystrophie, une kératodermie palmoplantaire, des ectropions, et enfin une alopécie.
Plusieurs types d’atteintes palmoplantaires s’observent au cours de ce syndrome, kératodermie essentiellement, mais aussi pustulose, ulcérations, bulles hémorragiques.
Comme dans le mycosis fongoïde, des lésions cliniques de parapsoriasis en grandes plaques, ou de papulose lymphomatoïde, peuvent précéder un syndrome de Sézary.
Avant la phase d’état, le tableau typique de syndrome de Sézary peut être précédé, pendant un temps plus ou moins long (quelques mois), par un état « prélymphomateux », durant lequel soit l’érythrodermie n’est pas complètement constituée, soit le nombre de cellules de Sézary est trop faible.
Les localisations viscérales sont, comme dans le mycosis fongoïde, le plus souvent asymptomatiques et découvertes à l’autopsie.
Cependant, Laroche et al, dans une étude prospective d’un groupe homogène de malades atteints de syndrome de Sézary, retrouvent, chez 42 % des malades, des manifestations neurologiques périphériques isolées.
C – CELLULE DE SÉZARY :
Recherchée sur les frottis sanguins colorés au May-Grünwald- Giemsa, c’est une cellule dont le noyau encoché possède une chromatine dense, parcourue de sillons en « coup d’ongle » dessinés à la surface nucléaire.
Sa description par Lutzner et Jordan, en 1968, en microscopie électronique, a ouvert l’ère du diagnostic ultrastructural des lymphomes cutanés épidermotropes à cellules T.
Ces cellules ont un aspect très caractéristique en microscopie électronique avec un noyau à contours très sinueux, profondément indenté, ce qui lui donne, en reconstruction tridimensionnelle, un aspect convoluté, cérébriforme.
Selon la taille de ces cellules, on distingue les grandes cellules de Sézary (> 12 ím) ou cellules de Sézary-Lutzner et les petites cellules de Sézary (£12 ím) ou cellules de Lutzner-Flandrin.
D – HISTOPATHOLOGIE :
Les caractéristiques histologiques cutanées sont similaires à celles du mycosis fongoïde bien que l’épidermotropisme soit fréquemment absent ou peu intense.
Les études récentes ayant cherché à comparer l’aspect anatomopathologique des lésions cutanées du mycosis fongoïde à celui des lésions de syndrome de Sézary ont abouti à des résultats discordants.
Ainsi, plusieurs auteurs ont montré que les seuls éléments qui différaient étaient la présence de « microabcès » de Pautrier dans le mycosis fongoïde et celle d’une acanthose dans le syndrome de Sézary ; une autre étude rapportait, au contraire, que la présence de « microabcès » de Pautrier et l’existence d’un oedème du derme superficiel étaient plus souvent observées dans le syndrome de Sézary, tandis qu’un épidermotropisme, une acanthose et une hyperkératose étaient plus souvent observés dans le mycosis fongoïde.
Les adénopathies envahies sont le siège d’un infiltrat dense de cellules de Sézary avec effacement total de l’architecture du ganglion.
Cet envahissement spécifique par les cellules atypiques est plus précoce dans le syndrome de Sézary que dans le mycosis fongoïde.
La moelle, en dépit de la présence dans le sang de nombreuses cellules de Sézary, est le plus souvent normale, la présence de quelques cellules pouvant traduire une simple contamination sanguine.
E – IMMUNOPHÉNOTYPE :
Il est similaire à celui décrit pour le mycosis fongoïde.
F – GÉNOTYPIE :
Un réarrangement monoclonal des gènes du TCR est retrouvé constamment dans le sang, si bien que la détection d’un clone cellulaire T dans le sang circulant peut être considérée comme un critère diagnostique de choix du syndrome de Sézary, permettant la différenciation avec les formes bénignes d’érythrodermie.
G – DIAGNOSTIC :
1- Diagnostic positif :
La démonstration d’un taux de 15 cellules de Sézary pour 100 lymphocytes circulants ou de 1000 cellules de Sézary par íL est un des critères classiques du diagnostic du syndrome de Sézary.
Le diagnostic peut également être retenu avec un nombre de cellules de Sézary moins important si le rapport CD4/CD8 des souspopulations lymphocytaires sanguines est supérieur ou au moins égal à 10.
Comme souligné au paragraphe précédent, le meilleur élément du diagnostic est la présence d’un clone circulant de cellules atypiques.
2- Diagnostic différentiel :
Les étiologies des érythrodermies sont variées et des cellules de Sézary ont été retrouvées, à la fois en microscopie optique et en microscopie électronique, dans le sang de nombreuses dermatoses bénignes, inflammatoires, ainsi que dans le mycosis fongoïde, mais en faible nombre.
Les pseudolymphomes actiniques peuvent évoluer vers une érythrodermie et s’accompagner d’un taux significatif de cellules de Sézary.
Le phénotype ordinairement CD8+ des cellules de Sézary circulantes est un bon argument pour le pseudolymphome actinique.
Les pseudosyndromes de Sézary d’origine médicamenteuse peuvent s’accompagner d’un nombre de cellules de Sézary identique à celui observé dans les syndromes de Sézary vrais.
Aussi, la recherche des médicaments pris par les patients doit-elle être systématique lors de toute suspicion de syndrome de Sézary.
H – PRONOSTIC :
Le taux de survie à 5 ans de 12 patients inclus dans le groupe néerlandais d’étude des lymphomes cutanés est de 11 %.
Les risques essentiels pour ces patients sont ceux de transformation de leur lymphome en un lymphome anaplasique, d’évolution fatale par dégradation de l’état général, d’une décompensation d’affections associées (insuffisance cardiaque…), d’une septicémie.
Dans le mycosis fongoïde érythrodermique et dans le syndrome de Sézary, les facteurs pronostiques importants sont l’âge des malades lors de la survenue de l’érythrodermie, le stade TNM de la maladie et l’existence ou non d’un envahissement sanguin.
Kim et al ont, en effet, montré que les patients de moins de 65 ans ont un pronostic plus favorable que ceux de 65 ans ou plus.
Un délai de plus de 10 ans avant le diagnostic s’associe à un pronostic plus favorable.
Le stade ganglionnaire (N de TNM) est corrélé de façon significative avec le taux de survie : les malades de stade III ont un pronostic plus favorable que ceux de stade IV.
Enfin, les malades ayant un syndrome de Sézary ont un plus mauvais pronostic que ceux ayant un mycosis fongoïde érythrodermique avec peu ou pas de cellules de Sézary circulantes (nombre de cellules de Sézary < 5 % des lymphocytes circulants).
I – TRAITEMENT :
L’évaluation des résultats thérapeutiques est en grande partie gênée par la sélection des patients, puisqu’il n’y a pas, à l’heure actuelle, de consensus sur les critères diagnostiques stricts du syndrome de Sézary.
1- Moyens thérapeutiques :
Étant donné l’extension systémique (ganglionnaire et sanguine) du syndrome de Sézary, les traitements à visée externe ne peuvent être envisagés que comme traitements associés.
Le méthotrexate, le chlorambucil et la prednisone combinés, l’interféron, la cytaphérèse, et la photophérèse extracorporelle soit utilisés seuls, soit associés à d’autres modalités de traitement, sont les traitements les plus utilisés.
La pentostatine est d’utilisation plus récente.
Le méthotrexate est prescrit à une posologie hebdomadaire de 15 à 50 mg, le chlorambucil à une posologie de 6 mg/j, associé à la prednisone à une posologie de 15 à 20 mg/j.
La photochimiothérapie extracorporelle, qui consiste en l’irradiation UVA ex vivo des lymphocytes mis en présence d’un psoralène, est une technique lourde et coûteuse, qui nécessite un appareil de photochimiothérapie.
Introduite en 1987 par Edelson, cette thérapeutique est pratiquement dépourvue d’effets secondaires graves en dehors du risque septique lié à la manipulation des cathéters veineux.
La nécessité d’une voie veineuse à débit suffisant conditionne sa réalisation.
2- Indications thérapeutiques :
Dans les formes faiblement hyperleucocytaires, l’association chlorambucil-prednisone, l’interféron et le méthotrexate à faibles doses sont les plus utilisés.
L’association chlorambucil-prednisone permet d’obtenir une réponse dans plus de 90 % des cas.
Le méthotrexate permet d’obtenir environ 40 % de rémissions complètes et 20 % de rémissions partielles.
Les rémissions à long terme obtenues chez quelques malades avec la pentostatine demandent à être confirmées.
La photochimiothérapie extracorporelle est l’indication de choix dans les formes fortement hyperleucocytaires.
Alors que les études antérieures semblaient montrer que la photochimiothérapie extracorporelle augmente le taux de survie, une étude récente suggère qu’elle ne prolonge pas plus la survie des malades que les monochimiothérapies.
En cas d’échec des monochimiothérapies et de la photochimiothérapie extracorporelle, on a recours aux polychimiothérapies.
Chalazodermie granulomateuse :
A – DÉFINITION :
C’est une affection extrêmement rare.
Rapportée pour la première fois par Convit et al sous le nom de « dermohypodermite atrophique granulomateuse », elle était considérée comme une maladie auto-immune (le premier cas décrit était postvaccinal).
On doit son individualisation comme lymphome à Ackerman.
B – CLINIQUE :
Elle atteint principalement l’homme et débute insidieusement chez l’adolescent ou chez l’adulte d’âge moyen par de petites plaques rouge violacé, initialement fermes qui, en s’étendant, deviennent atrophiques au centre pour donner des zones de peau flasque caractéristiques qui pendent parfois de manière spectaculaire.
Les lésions prédominent aux plis axillaires et inguinocruraux, épargnant toujours le visage.
L’association à une maladie de Hodgkin est classique, retrouvée dans près d’un tiers des cas. Des cas de mycosis fongoïde associés ont également été rapportés.
C – HISTOPATHOLOGIE :
Typiquement, il existe un infiltrat granulomateux massif du derme et de l’hypoderme, détruisant le tissu élastique, constitué de petits lymphocytes à noyau modérément convoluté, avec épidermotropisme sous forme d’éléments isolés ou groupés en petits amas, de nombreuses cellules géantes multinucléées et de macrophages.
L’activité de phagocytose des fibres élastiques et des lymphocytes est intense.
D – IMMUNOPHÉNOTYPE :
Les cellules atypiques ont le même phénotype que celles du mycosis fongoïde classique : CD3+, CD4+, CD8–.
E – GÉNOTYPE :
Un réarrangement monoclonal du récepteur T a été démontré.
F – DIAGNOSTIC DIFFÉRENTIEL :
On ne confondra pas la chalazodermie granulomateuse avec des lésions d’anétodermie ou de cutis laxa ; en revanche, un mycosis fongoïde granulomateux peut être de diagnostic différentiel difficile sur le plan histologique.
Le mycosis fongoïde granulomateux a les mêmes caractéristiques cliniques et le même pronostic que le mycosis fongoïde classique.
Il en diffère par la présence d’une composante granulomateuse dans l’infiltrat dermique, laquelle peut varier de quelques cellules géantes à de grands granulomes tuberculoïdes.
Par rapport à la chalazodermie granulomateuse, les cellules géantes, dans le derme du mycosis fongoïde granulomateux, sont plus éparses, ont un nombre plus réduit de noyaux.
Les lymphocytes peuvent être plus larges, hyperconvolutés.
La destruction du tissu élastique, enfin, est bien moindre.
G – SURVIE :
Les taux de survie sont difficilement chiffrables, étant donné le peu de cas rapportés dans la littérature ; la plupart des malades semblent avoir une évolution indolente.
H – TRAITEMENT :
Il n’y a pas, à l’heure actuelle, de traitement efficace de cette affection.
La radiothérapie a été rapportée comme arrêtant la progression de la maladie.
L’excision chirurgicale est suivie de récidives au bout de quelques mois.
Plusieurs auteurs ont noté des réponses partielles après traitement par corticothérapie générale.
La chimiothérapie, évaluée tardivement du fait que cette affection a d’abord été considérée comme une maladie auto-immune, a donné, elle aussi, des réponses partielles.
L’interféron-alpha en périlésionnel serait efficace.
Conclusion :
Il n’y a pas eu, ces dernières années, beaucoup de progrès thérapeutiques, ce qui s’explique en partie par la difficulté à mettre en oeuvre des études contrôlées.
Cependant, les travaux de réflexion menés au sein de groupes coopératifs tels que le groupe français d’études des lymphomes cutanés ont conduit à une utilisation plus nuancée et plus graduelle des thérapeutiques.
Ainsi, on privilégie les traitements locaux dans le mycosis fongoïde et ses variantes, et les traitements systémiques dans le syndrome de Sézary.
Les perspectives d’avenir résident, non pas dans les traitements cytotoxiques de la cancérologie conventionnelle qui ont, globalement, démontré leurs limites, mais dans les « immunointerventions » visant à perturber les interactions entre les cellules malignes et les cellules de l’environnement cutané.
")






")



{kind=link}