



















Introduction :
Le corps thyroïde est présent chez tous les vertébrés à sang chaud et froid.
 Il n’a acquis son statut de glande qu’à la fin du XIXe siècle avec l’évaluation des conséquences de la thyroïdectomie sur le développement normal des animaux.
Il n’a acquis son statut de glande qu’à la fin du XIXe siècle avec l’évaluation des conséquences de la thyroïdectomie sur le développement normal des animaux.
Les amphibiens ont constitué alors des modèles expérimentaux de choix, leur métamorphose (le passage de l’état larvaire à l’état adulte) étant sous la dépendance stricte des hormones thyroïdiennes (HT).
Le principe actif d’origine thyroïdienne, la thyroxine, fut isolé en 1925 par EC Kendall, qui obtint le prix Nobel en 1950.
Il est remarquable que la nature de cette sécrétion soit invariable selon les espèces : thyroxine et tri-iodo-thyronine sont produites par les glandes thyroïdes de tous les tétrapodes.
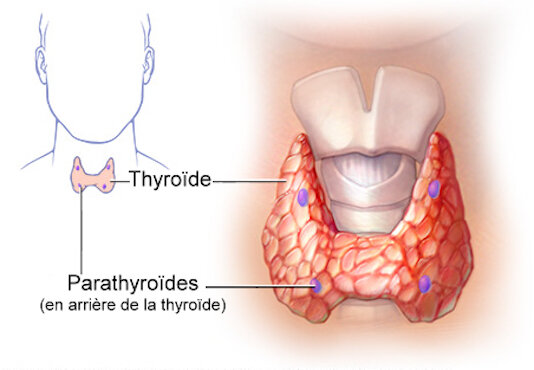
Rappels d’anatomie, d’embryologie et d’histologie :
A – ANATOMIE THYROÏDIENNE ET EXAMEN CLINIQUE :
La glande thyroïde est un corps impair et médian appliqué sur la partie antérieure de l’axe laryngotrachéal.
Elle présente une partie moyenne, mince et étroite, appelée isthme, et deux parties latérales volumineuses, les lobes droit et gauche.
Du bord supérieur de l’isthme part inconstamment un prolongement supérieur, le plus souvent latéralisé à gauche, de hauteur variable (au maximum jusqu’à l’os hyoïde) : le lobe pyramidal (ou pyramide de Lalouette).
Ses dimensions, variables selon les individus, sont approximativement de 5 cm de large (à la partie moyenne des deux lobes) et de 5 cm de haut (pour chaque lobe).
L’épaisseur est d’environ 1,5 cm.
Ces dimensions sont significativement plus importantes chez la femme que chez l’homme.
Le volume total de la glande est de l’ordre de 10 à 28 mL, son poids d’environ 30 g.
Située au tiers inférieur du cou, elle est maintenue par sa capsule fibreuse et surtout des adhérences à la trachée (ligaments thyrotrachéaux de Gruber) et à la gaine carotidienne (ligament latéral de Berry).
En position habituelle, elle se place en avant des deuxième et troisième anneaux trachéaux.
La palpation de la glande thyroïde se fait idéalement en se positionnant derrière le sujet assis et adossé.
Les doigts sont placés sous l’os hyoïde et glissent progressivement vers le bas pour repérer successivement l’incisure supérieure du cartilage thyroïde, la proéminence laryngée (pomme d’Adam), l’incisure inférieure du cartilage thyroïde, la partie antérieure de l’arc du cartilage cricoïde, puis le parenchyme thyroïdien lui-même.
En situation normale, il est difficile d’en apprécier les contours et la consistance exacte en raison de l’épaisseur des muscles préthyroïdiens (muscles sous- ou infrahyoïdiens).
Quelques éléments permettent toutefois d’évaluer l’existence d’un goitre : la palpation de la pyramide de Lalouette est toujours pathologique, le pôle supérieur des lobes ne se projette habituellement pas plus haut que la proéminence laryngée tandis que les pôles inférieurs descendent jusqu’au niveau du troisième ou du quatrième anneau trachéal.
Des pôles inférieurs non perceptibles alors que la glande thyroïde est en position cervicale moyenne sont très évocateurs d’un goitre plongeant dans le médiastin (attention cependant à la position basse de certaines glandes thyroïdes ptôsées).
Plus généralement encore, on parle d’hypertrophie thyroïdienne lorsque la hauteur d’un lobe thyroïdien dépasse la longueur de la deuxième phalange du pouce du sujet examiné (critère Organisation mondiale de la santé [OMS] dans les enquêtes épidémiologiques).
L’examen clinique de la thyroïde souligne également son caractère sensible ou douloureux, recherche un frémissement cataire (thrill) ou un souffle systolique, tous deux témoignant d’une hypervascularisation pathologique, et la présence d’adénopathies cervicales (jugulocarotidiennes, sous-mandibulaires, mastoïdiennes, occipitales et plus rarement sus-isthmiques).
B – EMBRYOLOGIE THYROÏDIENNE ET CONTRÔLE MOLÉCULAIRE DE L’ONTOGENÈSE THYROÏDIENNE :
Chez tous les tétrapodes, la glande thyroïde dérive d’une ébauche centrale et d’une paire d’ébauches latérales : les corps ultimobranchiaux.
L’ébauche centrale apparaît au début de la troisième semaine de développement (embryon de 2 cm) sous la forme d’un épaississement endodermique médian sur le plancher pharyngien.
Progressivement appendue à une invagination issue de cet épaississement (le canal thyréoglosse qui se résorbera au moins partiellement par la suite), l’ébauche thyroïdienne augmente de volume, devient bilobée et, du fait de l’allongement du cou de l’embryon, semble descendre vers sa position prélaryngotrachéale définitive.
À la septième semaine, les corps ultimobranchiaux, issus des quatrièmes poches pharyngées, se développent pour venir au contact des lobes latéraux de l’ébauche centrale avec lesquels ils fusionnent en se détachant du pharynx.
Ces ébauches latérales apportent au corps thyroïde des cellules neuroectodermiques, originaires des crêtes neurales qui, lors de cette fusion, envahissent les lobes thyroïdiens, s’éparpillent dans les follicules thyroïdiens en cours de formation et se différencient en cellules claires (cellules C ou parafolliculaires) productrices de calcitonine.
L’organogenèse et l’histogenèse thyroïdiennes sont sous la dépendance de mécanismes moléculaires complexes encore très imparfaitement compris.
Les avancées décisives concernent l’identification des facteurs de transcription spécifiques de la thyroïde (ou TTF).
Initialement isolés sur leur capacité à lier les séquences régulatrices des gènes codant des protéines spécifiques des cellules folliculaires de la thyroïde (comme le gène du récepteur de la thyroid stimulating hormone [TSH]), les TTF se sont révélés, dans des modèles murins d’inactivation génique (knockout), des acteurs essentiels de la migration et du développement de l’ébauche thyroïdienne.
Ils sont au moins au nombre de quatre et incluent les facteurs TTF1 (le premier exprimé dans l’ébauche thyroïdienne, également impliqué dans l’organogenèse pulmonaire), TTF2 (un facteur peut-être spécifiquement impliqué dans la migration thyroïdienne), Pax8 (indispensable à la différenciation des cellules endodermiques en cellules folliculaires) et enfin Hex (facteur impliqué plus généralement dans l’organogenèse de tous les dérivés de l’intestin pharyngien).
Chez les souris dépourvues de l’un ou l’autre de ces facteurs, la thyroïde est absente ou fortement hypoplasique, éventuellement ectopique.
C – NOTIONS D’HISTOLOGIE :
L’unité fonctionnelle de la thyroïde est le follicule thyroïdien, sphère de 200 à 300 µm de diamètre constituée d’une paroi épithéliale et d’un contenu amorphe, pâteux et jaunâtre à l’état frais : la colloïde.
L’épithélium est unistratifié et contient des cellules folliculaires, majoritaires, et des cellules plus claires, dites parafolliculaires.
Les cellules folliculaires sont encore dites vésiculaires ou appelées thyrocytes.
Le pôle basal des cellules folliculaires repose sur la lame basale du follicule, en contact avec les capillaires, alors que leur pôle apical, recouvert de microvillosités, se projette dans la colloïde.
Le noyau est central, d’autant plus basal que la cellule est active.
Les autres organites incluent des mitochondries, un réticulum endoplasmique granulaire, des ribosomes, un appareil de Golgi et de nombreuses vésicules d’exocytose et d’endocytose.
Les organites sont d’autant plus développés et la colloïde réduite que la glande est hyperactive.
Les cellules parafolliculaires représentent moins de 1 % du parenchyme thyroïdien total.
Elles sont plaquées contre la lame basale qui limite le follicule thyroïdien, et n’entrent jamais en contact avec la colloïde.
Elles sont caractérisées par la présence de grains de sécrétion, visibles en microscopie électronique, contenant la calcitonine.
Effets des hormones thyroïdiennes :
Le caractère invariable de la sécrétion thyroïdienne au cours de l’évolution suggère une fonction fondamentale pour les HT. De fait, les déficits ou les excès en HT perturbent le fonctionnement de multiples organes et systèmes organiques, chez l’enfant en développement comme chez l’adulte.
Les effets des HT sont variés mais s’exercent sans véritables organes cibles spécifiques.
Toute classification apparaît réductrice et artificielle même si, classiquement, on sépare les effets au cours du développement embryonnaire et foetal, les effets métaboliques et les effets spécifiques d’organe.
A – RÔLE DANS LE DÉVELOPPEMENT EMBRYONNAIRE ET FOETAL :
Les besoins en HT existent probablement très précocement au cours de la vie intra-utérine ; ils sont initialement assouvis par la production maternelle puisque les hormones libres sont capables de traverser le placenta.
La thyroïde de l’embryon devient elle-même fonctionnelle vers la dixième semaine de développement, se substituant alors à la thyroïde maternelle.
Chez l’homme, les conséquences d’un déficit embryonnaire ou foetal en HT se remarquent essentiellement au niveau du squelette et du système nerveux, même si les modèles murins d’inactivation génique supportent leur rôle fondamental dans la différenciation d’autres systèmes dont surtout le tractus digestif.
Pour l’os, elles apparaissent plus nécessaires à l’ossification qu’à la croissance : les enfants déficitaires ont un poids et une taille dans les limites de la normale mais leurs épiphyses osseuses sont peu ou pas calcifiées.
Pour le système nerveux, l’appréciation d’un déficit intra-utérin est difficilement perceptible à la naissance puisque la maturation nerveuse est alors loin d’être achevée.
De façon générale, on peut signaler que les HT jouent un rôle fondamental à la fois dans la différenciation (développement des axones et des dendrites) et la migration neuronales, la différenciation gliale (myélinisation des fibres nerveuses) et la synaptogenèse.
Si le retard d’ossification peut être rattrapé secondairement par un traitement substitutif adéquat, le retard de maturation nerveuse est plus difficilement corrigeable, soulignant l’importance d’une recherche systématique de l’hypothyroïdie lors des échographies foetales, du dépistage néonatal et, de façon plus générale, d’un diagnostic et d’un traitement les plus précoces possibles.
B – EFFETS MÉTABOLIQUES DES HORMONES THYROÏDIENNES :
L’action générale des HT est d’accroître les métabolismes : une augmentation du métabolisme de base est décelée au cours des hyperthyroïdies.
Les HT augmentent en effet la consommation d’oxygène de tous les tissus et la production de chaleur par l’organisme en favorisant la thermogenèse inhérente aux réactions métaboliques (thermogenèse obligatoire), notamment les réactions mitochondriales qui utilisent l’adénosine triphosphate (ATP) comme substrat.
Elles sont hyperglycémiantes en accélérant l’absorption intestinale de glucose, en accroissant la glycogénolyse et en réduisant la glucogenèse et la néoglucogenèse d’origine protidique ou lipidique.
Elles stimulent également l’utilisation cellulaire de glucose.
Dans le métabolisme protidique, les HT interviennent de façon discordante, avec une stimulation conjointe de la synthèse et du catabolisme protidiques.
Ce dernier prédomine cependant avec une fonte musculaire et une augmentation consécutive de la créatininurie remarquables dans les hyperthyroïdies (négativité de la balance azotée).
L’action des HT sur le métabolisme lipidique est également complexe avec une action stimulatrice de la synthèse du cholestérol aux concentrations physiologiques mais inhibitrice à des concentrations supérieures.
La baisse du cholestérol total et du cholestérol low density lipoprotein (LDL) en particulier constitue un marqueur classique de l’hyperthyroïdie.
Les HT augmentent la cétogenèse et l’absorption intestinale du calcium.
Sur l’os constitué, elles ont des effets contrastés, associant destruction et synthèse osseuses, ce qu’exprime bien l’augmentation de l’ensemble des marqueurs du remodelage osseux sous leur influence.
L’action ostéolytique prédomine cependant, expliquant l’ostéoporose et la diminution de la densité osseuse observées dans l’hyperthyroïdie prolongée.
C – EFFETS SPÉCIFIQUES D’ORGANES :
Après la naissance, les HT participent encore à la différenciation osseuse.
Elles stimulent la chondrogenèse, la croissance des cartilages de conjugaison et l’ossification enchondrale (ossification à partir du cartilage).
Si le déficit se poursuit en période néonatale, le retard de croissance finit par se déclarer, résultant probablement de l’absence de leur effet stimulant physiologique sur les productions hypophysaires de l’hormone de croissance (growth hormone : GH) et hépatique de l’insulin-like growth factor-1 (IGF-1).
Comme les autres muscles, le myocarde est sensible à l’action des HT qui ont des effets chronotrope (accélération du rythme cardiaque), inotrope (augmentation de la contractilité), dromotrope (amélioration de la conduction) et lusitrope (accélération de la relaxation ventriculaire).
En périphérie, les HT diminuent les résistances vasculaires en relâchant les muscles lisses.
Les résultantes sont l’augmentation du débit cardiaque et l’hypertrophie ventriculaire.
Ces effets miment les actions b-adrénergiques des catécholamines et de fait, les HT semblent agir au moins partiellement en favorisant l’effet de ces substances.
Leurs effets cardiaques sont d’ailleurs contrecarrés par les b-bloquants.
Enfin, les HT stimulent la motilité intestinale et accélèrent le transit digestif.
Biosynthèse des hormones thyroïdiennes :
A – STRUCTURE DES HORMONES THYROÏDIENNES :
Les hormones produites par la glande thyroïde sont dérivées de la forme lévogyre (L) d’un acide aminé, la tyrosine, et sont donc caractérisées par la présence des groupements acide (COOH) et amine primaire (NH2).
Elles contiennent également deux noyaux phénols, appelés anneaux interne et externe.
Les six atomes de carbone qui constituent ces noyaux sont numérotés de 1 à 6 (ou 1’ à 6’ dans l’anneau externe) dans le sens antihoraire. Sur les anneaux sont branchés trois ou quatre atomes d’iode.
Sont ainsi produites la thyroxine (ou T4 ou 3, 5, 3’, 5’ tétra-iodo-thyronine) et la 3, 5, 3’ tri-iodo-thyronine (ou T3).
La T3 n’est produite par la thyroïde qu’en quantité réduite (20 %).
Elle provient essentiellement de la désiodation de l’anneau externe de la T4 par les tissus cibles périphériques (foie, rein, muscle, cerveau), cette production périphérique s’adaptant aux conditions physiologiques.
B – GRANDES ÉTAPES DE LA BIOSYNTHÈSE DES HORMONES THYROÏDIENNES :
Les grandes étapes de la biosynthèse des HT comprennent :
– toutes les étapes qui permettent la mise à disposition de l’iode pour son incorporation dans les précurseurs des HT : le transfert de l’iode capté dans le sang circulant à travers la membrane cellulaire du pôle basal de la cellule folliculaire (rôle d’un transporteur spécifique : le symporteur sodium-iodure), son transfert intracellulaire vers le pôle apical, son transfert à travers la membrane cellulaire du pôle apical (rôle d’un transporteur spécifique : la pendrine) ;
– toutes les étapes qui permettent son incorporation dans la thyroglobuline, une protéine spécifique qui contient les précurseurs des HT.
Cette phase, dite d’organification de l’iodure, nécessite une oxydation enzymatique de l’iode, utilisant des enzymes spécifiques catalysant la réaction (thyroperoxydase) ou générant les matériaux qui lui sont nécessaires (système générateur de peroxyde d’hydrogène ou H2O2) ;
– toutes les étapes qui libèrent les HT de la thyroglobuline et leur libération dans le sang après transfert de la solution de stockage, la colloïde, vers le pôle basal de la cellule.
C – CAPTAGE DE L’IODURE :
1- Origines de l’iodure :
Traditionnellement, l’organisme puise l’iode dont il a besoin dans les aliments issus de la mer.
L’eau de mer est en effet la principale source d’iode ; elle en contient jusqu’à 5 parties par million.
Le sel de table, lorsqu’il est enrichi en iode, constitue la source alimentaire la plus simple et la plus efficace pour accroître l’apport iodé dans les régions déficitaires.
En France, l’apport moyen d’iodure est de l’ordre de 50 à 100 µg/j dans les conditions alimentaires normales, ce qui est considéré comme la limite de la carence iodée.
L’iodure peut également être apporté par l’administration de médicaments ou de produits de contraste radiologiques ou encore l’application de produits antiseptiques.
Une surcharge en iode peut apparaître, susceptible d’induire à la fois des hyperthyroïdies et des hypothyroïdies.
Il existe enfin une production d’iode endogène, liée à la désiodation périphérique et intrathyroïdienne des HT et de leurs catabolites.
Dans l’organisme, l’iode ainsi disponible se répartit dans un espace de diffusion qui correspond à environ 35 % du poids corporel et comprend la thyroïde (iode organique), les glandes salivaires, gastriques et mammaires, le secteur vasculaire et les organes d’élimination (iode inorganique).
Si de l’iode est présent dans les selles ou la sueur, le rein est le principal émonctoire avec une clairance quotidienne de l’ordre de 30 mL/min.
À l’équilibre, la quantité d’iodure excrétée égale la quantité ingérée et la mesure de l’iodurie des 24 heures constitue une bonne estimation des apports (alimentaires et/ou iatrogènes).
La concentration sérique d’iodure, résultant des apports et de l’élimination, oscille normalement entre 0,1 et 0,3 µg/100 mL.
2- Transport de l’iode au pôle basolatéral : symporteur du sodium et de l’iodure
C’est sous la forme d’un ion (iodure) que l’iode est activement capté au pôle basolatéral des cellules folliculaires : il ne s’agit pas d’une entrée passive.
Ce transport actif est saturable et réversible.
Une autre particularité remarquable est son adaptabilité aux fluctuations d’apport de l’iodure : l’entrée d’iode est stable malgré un apport iodé accru ou réduit.
Enfin, l’influx d’iodure dans la cellule est inhibé de façon compétitive par d’autres anions comme les ions perchlorate (ClO4 -), pertechnétate (99mTcO4 -), thiocyanate (SCN-) et perrhenate (ReO4-).
Cette constatation est à la base du test au perchlorate qui révèle l’efflux passif de l’iode non organifié.
Il est considéré comme positif lorsque l’on constate après 1 heure une diminution d’au moins 10 % du taux de fixation de l’iode radioactif.
Cette diminution importante signifie que l’iode n’a pas été organifié en quantités suffisantes et ne peut suivre les étapes ultérieures de la biosynthèse hormonale.
Ces données sont connues depuis longtemps mais ce n’est que récemment que la « pompe à iodure » a été identifiée au niveau moléculaire, d’abord chez le rat puis chez l’homme.
Chez l’homme, il s’agit d’une protéine membranaire glycosylée de 643 acides aminés, dont le gène est situé sur le chromosome 19.
L’analyse de sa structure lui prédit 13 domaines transmembranaires.
Le transport de l’iodure aboutit à un gradient de concentration entre milieux intra- et extracellulaires d’environ 30.
La force motrice nécessaire à ce transport actif contre gradient utilise un flux entrant concomitant de Na+, d’où le nom de symporteur du sodium et de l’iodure (ou NIS).
Le gradient de Na+ est assuré par une enzyme consommant de l’ATP : une ATPase dépendante du Na+ et du K+ (Na+ /K+ ATPase) dont l’inhibition par l’ouabaïne abolit le transport de l’iodure.
Dans la thyroïde, le NIS est exprimé spécifiquement dans la membrane basolatérale, mais une forte expression est également détectée dans d’autres tissus capables de concentrer l’iode comme les glandes salivaires, gastriques ou mammaires.
L’expression mammaire est maximale en fin de grossesse et au cours de l’allaitement ; elle assure une concentration forte d’ion iodure dans le lait permettant la synthèse d’HT par le nourrisson.
Une expression plus faible a également été notée dans d’autres tissus qui ne concentrent pas l’iode comme l’hypophyse, le pancréas, les gonades, la prostate, la surrénale ou le thymus.
La véritable capacité à concentrer l’iode dépendrait de la possibilité d’une stimulation de l’activité du NIS et en particulier de la présence d’un récepteur de la TSH : la TSH augmenterait à la fois l’expression et l’activité de transport du NIS.
L’expression du NIS est également sous la dépendance des facteurs de transcription spécifiques de la thyroïde : les facteurs TTF1, TTF2 et Pax8.
En complément de son rôle dans la physiologie thyroïdienne, le captage de l’iode par le NIS constitue un prérequis crucial pour l’imagerie fonctionnelle de la glande lors des scintigraphies diagnostiques et le traitement radioisotopique des affections thyroïdiennes bénignes et malignes.
Les implications du NIS en pathologie sont rares.
Des cas de déficit congénital en transport de l’iodure ont été sporadiquement rapportés.
La transmission de ce déficit est autosomique récessive et une consanguinité est fréquente dans la famille.
Ce déficit s’accompagne d’un goitre congénital, de l’incapacité à concentrer l’iodure et, quand il est complet, d’une hypothyroïdie.
Dans toutes les familles testées, une anomalie des deux allèles du gène codant le NIS a été découverte. L’étude phénotypique de ces familles a été très instructive pour comprendre le fonctionnement du NIS.
La remarque la plus intéressante est que le phénotype est variable d’un sujet atteint à l’autre ; des patients hétérozygotes pour la mutation peuvent présenter des anomalies cliniques mineures (goitre et hypothyroïdie fruste).
Ces données suggèrent l’existence de cofacteurs du NIS, pour l’instant non identifiés.
D – TRANSPORT TRANSMEMBRANAIRE DE L’IODURE AU PÔLE APICAL : LA PENDRINE
L’iodure entré dans la cellule folliculaire peut diffuser vers l’extérieur (probablement passivement du fait du gradient électrochimique) ou être transféré dans la lumière folliculaire et la colloïde.
Longtemps considéré comme un transport passif, le transport transmembranaire de l’iodure au pôle apical nécessite en fait un transporteur protéique actif qui n’a que récemment été identifié au niveau moléculaire : la pendrine.
Il s’agit du produit du gène PenDred’s Syndrome (PDS), présent sur le chromosome 7, dont les mutations, à l’état homozygote, expliquent l’apparition du syndrome de Pendred, ce syndrome rare associant surdité congénitale, goitre survenant dans l’enfance et hypothyroïdie d’intensité variable.
La pendrine est une protéine transmembranaire de 780 acides aminés et 86 kDa dont la structure prédite comporte 11 domaines transmembranaires.
Dans la thyroïde, elle est spécifiquement exprimée au pôle apical des thyrocytes, avec une intensité variable d’un thyrocyte à l’autre et d’un follicule à l’autre.
Elle est également exprimée dans le rein, le cerveau foetal, le placenta et la membrane labyrinthique de l’oreille interne.
La pendrine se comporte comme un transporteur des ions iodure et chlorure selon un mécanisme qui reste à évaluer précisément.
Contrairement à l’activité du NIS, ce transport apparaît indépendant de la TSH, de la concentration en Na+ ou de l’apport iodé.
Sur le plan physiopathologique, dans le syndrome de Pendred, l’altération fonctionnelle de ce transport serait responsable, dans la thyroïde, du défaut d’incorporation de l’iode dans la thyroglobuline (à l’origine du goitre et de l’hypothyroïdie) et, dans l’oreille interne, d’une anomalie hydraulique des liquides labyrinthiques (à l’origine de la surdité par dilatation des sacs et des canaux endolymphatiques puis des aqueducs vestibulaires, visibles lors de l’exploration morphologique des rochers temporaux).
Plus de 50 anomalies différentes du gène codant la pendrine ont été publiées chez des patients atteints de syndrome de Pendred.
Il faut noter là encore une certaine variabilité phénotypique avec la description de mutations homozygotes du gène PDS chez des patients porteurs d’une surdité congénitale avec dilatation des aqueducs vestibulaires sans anomalie aucune de la fonction thyroïdienne.
On pressent ainsi l’activité conjointe à celle de la pendrine d’autres facteurs intervenant dans la mise à disposition de l’iode à la thyroglobuline dans la colloïde. Récemment, un autre transporteur apical de l’iode a été décrit.
Appelé apical iodide transporter (AIT), il a été identifié sur la base de son identité avec le NIS et favoriserait la diffusion passive de l’iode à travers la membrane apicale du thyrocyte où il est exprimé.
E – ORGANIFICATION DE L’IODURE ET SYNTHÈSE HORMONALE :
1- Présentation globale :
L’iodure capté par la cellule folliculaire et excrété dans la colloïde est incorporé à la thyroglobuline, qui constitue le support essentiel de la biosynthèse des HT.
Cette incorporation est appelée organification de l’iode et l’iode est dit alors organique.
La thyroglobuline est synthétisée dans la cellule folliculaire et excrétée dans la colloïde.
Sous l’action d’une peroxydase spécifique, la thyroperoxydase ou TPO, l’iode est couplé à certains résidus transformés de tyrosine présents dans la thyroglobuline.
Dans cette réaction, le peroxyde d’hydrogène H2O2 est un élément limitant et sa mise à disposition dépend d’un système enzymatique : le système générateur d’H2O2.
La thyroglobuline iodée s’accumule dans la colloïde, assurant ainsi un stockage des HT sous la forme d’une véritable prohormone inactive, dans un espace clos isolé des influences métaboliques non spécifiques.
Sous la stimulation de facteurs spécifiques, la thyroglobuline sera réabsorbée par la cellule folliculaire, clivée dans des vésicules lysosomiales de façon à libérer les HT.
2- Thyroglobuline :
La thyroglobuline est une protéine spécifiquement produite par la glande thyroïde.
Cette particularité est utilisé en pratique clinique dans les surveillances des cancers thyroïdiens différenciés après exérèse chirurgicale : une réascension des taux de thyroglobuline signe une récidive ou une persistance tumorale.
La thyroglobuline se présente sous la forme d’une protéine homodimérique de 660 kDa à la fois glycosylée, phosphorylée et sulfatée.
Elle contient deux sous-unités identiques comportant chacune 2 749 acides aminés.
Sa capacité d’iodation dépend de la présence, dans sa structure protéique, de 134 résidus tyrosine, dont seulement quelques-uns (5 à 16) participent réellement à la synthèse hormonale.
L’iodation des résidus tyrosine aboutit à la formation d’iodotyrosines (mono- ou di-iodo-tyrosines) et le couplage de deux iodotyrosines à la formation des iodothyronines (T3 ou T4).
Les résidus tyrosine qui supportent les HT formées ou en formation (sites accepteurs) sont, pour ceux qui ont été localisés, situés aux extrémités de la thyroglobuline.
Ils portent toujours une di-iodo-tyrosine tandis que les sites donneurs offrent souvent une di-iodo-tyrosine et plus rarement une mono-iodo-tyrosine ; ceci explique la sécrétion préférentielle de T4 par rapport à la T3.
Le résidu situé en position 5 supporte la majorité de la synthèse hormonale, jusqu’à 70 % dans certaines espèces ; il se couple spécifiquement avec le résidu tyrosine en position 130 (site donneur).
Enfin, la thyroglobuline est une protéine fortement antigénique : elle est le principal autoantigène thyroïdien et au moins 40 déterminants antigéniques ont été répertoriés.
Le gène de la thyroglobuline est situé sur le chromosome 8. Sa particularité essentielle est la longueur de la région qu’il couvre : jusqu’à 300 000 paires de bases !
Il comprend 42 exons et des séquences régulatrices complexes qui portent des éléments de réponse pour des facteurs de transcription spécifiques de la thyroïde.
Les facteurs TTF1 et Pax8 stimulent son expression alors que les facteurs TTF2 et Hex l’inhibent.
La thyroglobuline, a retro, contrôlerait probablement indirectement l’expression de protéines spécifiques de la cellule folliculaire comme TTF1, réalisant ainsi une véritable boucle de régulation.
La TSH, mais aussi l’IGF-1 stimulent indirectement (par une voie utilisant leurs récepteurs membranaires) l’expression du gène de la thyroglobuline.
Des hypothyroïdies génétiquement déterminées ont été rapportées dans lesquelles un défaut qualitatif ou quantitatif de production de la thyroglobuline est spécifiquement présent.
Chez l’homme, quelques mutations du gène de la thyroglobuline ont été identifiées, habituellement portées à l’état homozygote, conduisant à un codon stop prématuré et une thyroglobuline tronquée, ou à une anomalie du processus complexe d’épissage de l’acide ribonucléique messager (ARNm).
Dans d’autres cas, des mutations de séquences spécifiques entraînent un défaut de sécrétion par anomalie d’adressage intracellulaire de la thyroglobuline.
Dans d’autres familles avec goitre, le gène de la thyroglobuline est normal et la thyroglobuline normalement glycosylée mais la thyroglobuline ne se replie pas correctement, s’accumulant dans le réticulum endoplasmique.
L’anomalie d’une enzyme impliquée spécifiquement dans les processus de maturation de la thyroglobuline est l’hypothèse retenue.
3- Thyroperoxydase :
La peroxydase thyroïdienne ou thyroperoxydase est une enzyme majeure de la biosynthèse hormonale thyroïdienne.
Elle est responsable de l’oxydation de l’iodure, de son incorporation ultérieure dans la thyroglobuline et du couplage des iodotyrosines en iodothyronines.
Ces activités dépendent étroitement d’un substrat, l’H2O2, qui lui est fourni par un système enzymatique spécifique.
La TPO est une glycoprotéine membranaire de 933 acides aminés et localisée au pôle apical de le cellule folliculaire.
Son extrémité C-terminale contient une région d’ancrage à la membrane tandis que sa moitié N-terminale, qui sera libre dans la lumière folliculaire, supporte l’activité enzymatique. Un résidu histidine, situé en position 407, sert d’ancrage à la structure hémique : la TPO, comme les autres peroxydases, est une hémoprotéine.
Comme la thyroglobuline, la TPO est antigénique ; les anticorps anti-TPO correspondent aux anticorps antimicrosomes.
Le gène de la TPO est localisé sur le chromosome 2. Il s’étend sur plus de 150 000 paires de base et comprend 17 exons dont l’exon 10 (171 nucléotides) est soumis à un épissage alternatif.
Il existe donc deux isoformes de TPO dont l’une est dépourvue de l’exon 10.
L’expression du gène est strictement limitée à la cellule folliculaire de la thyroïde.
Elle est stimulée par la TSH via son récepteur membranaire.
Les facteurs transcriptionnels qui véhiculent le signal stimulant issu de la TSH sont peut-être les facteurs TTF1, TTF2 et Pax8 qui reconnaissent des éléments de réponse spécifiques situés dans le promoteur et l’enhancer du gène.
Des mutations homozygotes du gène de la TPO ont été identifiées dans des familles d’hypothyroïdie congénitale avec goitre.
Elles s’accompagnent d’un défaut d’organification de l’iodure qui peut être détecté par le test au perchlorate.
Elles sont transmises sur un mode autosomique récessif.
Les anomalies du gène de la TPO représenteraient les erreurs congénitales les plus fréquentes du métabolisme des HT.
4- Système générateur d’H2O2 :
L’activité catalytique de la TPO dépend strictement de l’oxydation de son hème par une molécule de peroxyde d’hydrogène (H2O2) ; la TPO passe alors d’une forme inactive à une forme active appelée « composé I ».
Elle utilise de plus une molécule d’H2O2 à chaque fois qu’elle incorpore un atome d’iode sur une tyrosine ou qu’elle réalise un couplage d’iodotyrosines en iodothyronines.
Pour synthétiser la T4, la TPO a donc besoin de cinq molécules d’H2O2 ; la quantité d’H2O2 est un facteur limitant de la biosynthèse des HT et le système générateur d’H2O2 se doit d’être performant.
L’enzyme responsable est une nicotinamide-adénine-dinucléotide phosphate (NADPH) oxydase spécifique de la thyroïde appelée Thyroid OXydase (TOX) ou THyroid OXydase (THOX).
Présente à la membrane apicale de la cellule folliculaire, elle produit directement l’H2O2 à partir du NADPH, de l’oxygène et d’un ion hydrogène :
NADPH + O2 + H+ -> NADP+ + H2O2
Il existe en fait deux THOX, l’une de 1 548 acides aminés (THOX2 ou p138tox), l’autre de 1 551 résidus (THOX1), codées par des gènes différents localisés cependant conjointement sur le bras long du chromosome 15.
Les deux protéines sont identiques à 83 %, probablement transmembranaires et toutes deux des flavoprotéines, capables de fixer le flavine adénine nucléotide (FAD) nécessaire à leur activité enzymatique.
Le calcium est également un cofacteur nécessaire à l’activation enzymatique.
L’analyse de la structure primaire de la THOX2 prévoit une organisation en sept domaines transmembranaires ou en tout cas fortement hydrophobes.
Les domaines de liaison du FAD et du NADPH seraient situés à l’extrémité C-terminale (intracellulaire) tandis que le domaine de liaison du calcium, lui aussi intracellulaire, serait situé entre les premier et deuxième segments transmembranaires.
Une étude plus récente montre que les THOX sont faiblement exprimées à la membrane cellulaire et suggère que d’autres protéines enzymatiques interviennent in vivo dans la génération des molécules d’H2O2.
Récemment, des anomalies du gène de la THOX2 ont été identifiées chez des patients atteints d’hypothyroïdie congénitale avec déficit de l’organification de l’iode.
Les anomalies identifiées sont, soit homozygotes, aboutissant à une hypothyroïdie sévère et permanente, soit hétérozygotes, conduisant à une hypothyroïdie légère et transitoire.
5- Sécrétion des hormones thyroïdiennes :
La première phase de sécrétion des HT est la recapture de la thyroglobuline stockée dans la colloïde par la cellule folliculaire.
Cette étape utilise un procédé d’endocytose, ou plus précisément d’endopinocytose puisque le matériel récupéré est liquide.
Selon la taille de la gouttelette de colloïde récupérée, on parle de micropinocytose (vésicules de 100 nm de diamètre) ou de macropinocytose (gouttelettes jusqu’à 2 µm de diamètre, en moyenne 1,5 µm).
Comme la phagocytose, la macropinocytose utilise la formation de pseudopodes à partir de la membrane apicale, près des bords de la cellule, à proximité des jonctions cellulaires.
La formation des pseudopodes et des gouttelettes de colloïde est stimulée par la TSH.
Ce processus de macropinocytose ne serait pas sélectif : la thyroglobuline récupérée peut être non iodée ou n’avoir subi qu’une iodation partielle.
La micropinocytose de vésicules de colloïde constituerait en fait chez l’homme le mécanisme d’internalisation préférentiel de la thyroglobuline, en tout cas en situation normale (la macropinocytose pourrait être mise en jeu en cas de nécessité d’une production intense d’HT).
Comme la macropinocytose, la micropinocytose est stimulée par la TSH.
Ce processus nécessite non plus la projection de pseudopodes dans la colloïde, mais la formation d’une invagination, d’un puits dans la membrane plasmique.
Dans le cadre de la recapture de thyroglobuline, le puits se fait entre deux microvillosités et est recouvert, sur sa face cytoplasmique, d’une protéine spécifique de l’endocytose : la clathrine.
Le puits se recouvre, la couverture de clathrine se referme et permet à la vésicule ainsi formée de se détacher de la membrane cytoplasmique.
La vésicule recouverte se libère ensuite de la couverture de clathrine pour former une vésicule nue qui fusionne rapidement avec un organite intracellulaire accepteur appelé endosome précoce.
La molécule internalisée est ensuite transférée de proche en proche vers un endosome tardif (ou prélysosome) puis vers un lysosome.
La micropinocytose de la thyroglobuline serait un phénomène sélectif : après internalisation, la thyroglobuline non ou peu iodée serait réorientée vers la colloïde.
Les mécanismes sousjacents précis ne sont pas déterminés ; on pense que des récepteurs spécifiques, localisés dans la membrane des endosomes, reconnaissent le contenu riche en certains résidus sucrés (galactose, N-acétylglucosamine, N-acétylgalactosamine) et pauvre en acide sialique de la thyroglobuline non iodée.
Il s’agit par exemple du récepteur des asialoglycoprotéines qui présente une affinité maximale pour ses ligands dans un contexte de pH bas.
Par cette liaison forte à pH bas, ce récepteur permettrait à la thyroglobuline d’échapper à la protéolyse lysosomique (lors de la fusion des vésicules internalisées avec les lysosomes, les protéines dont les récepteurs ont une affinité maximale à pH neutre, se dissocient des récepteurs et sont protéolysées alors que les récepteurs à affinité maximale à pH acide ne se dissocient pas de leurs ligands et les empêchent d’être protéolysées).
De façon anecdotique, la présence du récepteur thyroïdien des asialoglycoprotéines est nécessaire à la répression de l’expression de plusieurs protéines spécifiques de la thyroïde (TTF1, TTF2, Pax-8, NIS) exercée par la thyroglobuline.
Les gouttelettes de colloïde ou les vésicules fusionnées aux endosomes fusionnent dans le cytoplasme cellulaire avec des lysosomes.
Dans ces compartiments subcellulaires, la thyroglobuline entre en contact avec des enzymes du lysosome qui libèrent les résidus hormonaux.
Les enzymes nécessaires à la protéolyse de la thyroglobuline sont des endopeptidases (qui fractionnent les protéines en peptides d’au moins cinq acides aminés en coupant des liaisons peptidiques internes) et des exopeptidases (qui réduisent les peptides en dipeptides ou en acides aminés en supprimant un à un les résidus des extrémités).
Les enzymes libérant les HT pourraient être différentes selon que les résidus hormonaux sont situés à l’extrémité N-terminale ou C-terminale de la thyroglobuline.
Une fois les HT libérées, la thyroglobuline serait dégradée entièrement par d’autres enzymes protéasiques mais aussi des enzymes capables de supprimer les chaînes d’hydrocarbones, de sulfates ou de phosphates.
Les mono-iodotyrosines et les di-iodo-tyrosines non utilisées pour la formation des HT sont également libérées ; elles sont rapidement décomposées en résidus tyrosine et en ions iodure par une désiodase spécifique localisée dans la membrane du réticulum endoplasmique.
T3 et T4 seraient, quant à elles, libérées dans le cytoplasme de la cellule folliculaire et, classiquement, diffuseraient passivement dans la circulation sanguine à travers la membrane cytoplasmique du pôle basolatéral.
Il est en fait vraisemblable, mais cela n’est pas démontré, que les HT utilisent aussi (surtout ?) des transporteurs membranaires et/ou sont conservées dans des vésicules qui s’ouvrent secondairement à la membrane cytoplasmique du pôle basolatéral par exocytose.
Certaines vésicules, voire certaines gouttelettes de colloïde, échappent à la fusion avec les lysosomes et parviennent à la membrane cytoplasmique du pôle basolatéral où elles s’ouvriraient par exocytose.
Ce mécanisme, appelé transcytose, explique la production de thyroglobuline intacte, plus ou moins iodée, dans la circulation générale, où elle sert de marqueur sérique de la présence d’un parenchyme thyroïdien fonctionnel.
Il serait sous la dépendance d’un autre récepteur apical de la thyroglobuline, la mégaline.
Il s’agit d’une glycoprotéine de 330 kDa, appartenant à la famille des récepteurs des LDL et se comportant comme un récepteur multiligand transmembranaire impliqué dans les processus d’endocytose.
Elle est exprimée à la surface de plusieurs cellules épithéliales polarisées (rein, intestin, glandes parathyroïdes, thyroïde…).
Dans la thyroïde, la mégaline est exprimée à la membrane apicale des thyrocytes.
Cette expression et son activité de liaison de la thyroglobuline sont stimulées par la TSH.
La liaison thyroglobuline-mégaline est inhibée par l’héparine et facilitée par les héparane-sulfate protéoglycanes.
L’expression de la mégaline est stimulée dans la maladie de Basedow, l’augmentation des taux sériques de thyroglobuline s’expliquant ainsi par le goitre mais aussi par une augmentation de la transcytose.
Des autoanticorps antimégaline sont produits au cours de cette maladie mais également dans d’autres maladies autoimmunes sans que l’on en connaisse encore la signification.
Chaque jour, la thyroïde produit de l’ordre de 85 à 125 µg de T4 (environ 110 nmol).
La T3 est produite en quantités beaucoup plus faibles ; seuls 20 % de la T3 produite chaque jour proviennent de la thyroïde, 80 % étant issus de la transformation à partir de la T4.
La concentration en HT des veines thyroïdiennes augmente dans les 30 minutes qui suivent une injection de TSH.
Cet effet découle principalement d’une stimulation de l’endocytose de la thyroglobuline et donc d’une libération partielle des stocks situés dans la colloïde.
La libération des HT dans les lysosomes et leur sécrétion proprement dite dans les capillaires folliculaires ne semblent pas sous le contrôle de la TSH.







")


")
{kind=link}