



















La sarcoïdose est une maladie inflammatoire granulomateuse diffuse de cause indéterminée.Elle se caractérise par un polymorphisme clinique lié à des atteintes polyviscérales, au premier rang desquelles sont les localisations médiastino-pulmonaires. L’unité de la maladie est liée à la présence dans les organes atteints de la lésion histologique élémentaire : le nodule sarcoïdosique. L’évolution de la sarcoïdose est dans la grande majorité des cas favorable. Néanmoins, des complications graves liées à certaines localisations viscérales (œil, cœur, système nerveux central) ou à l’évolution vers l’insuffisance respiratoire chronique du fait de la constitution d’une fibrose pulmonaire sont possibles et justifient donc une surveillance régulière des malades atteints, même si initialement il n’existe pas de critère de mise en place d’une corticothérapie.
DONNEES GENERALES
Définition :
 La sarcoïdose est une granulomatose systémique de cause inconnue, touchant avec prédilection la sphère médiastino-pulmonaire.
La sarcoïdose est une granulomatose systémique de cause inconnue, touchant avec prédilection la sphère médiastino-pulmonaire.
L’évolution est souvent favorable mais des complications respiratoires et extra-respiratoires variées sont possibles.
La prise en charge thérapeutique consiste tantôt en une surveillance simple, tantôt en une corticothérapie d’emblée ou secondaire.
Epidémiologie :
60% des cas de sarcoïdose sont découverts entre 20 et 40 ans.
La sarcoïdose est plus fréquente dans le sexe féminin en raison d’un deuxième pic d’incidence périménopausique.
Elle est plus fréquente et plus sévère chez les Noirs.
Anatomie pathologique :
Les granulomes constituent la lésion de base de la sarcoïdose. Ce sont des granulomes tuberculoïdes, sans nécrose, caractéristiques d’une réponse immunitaire de type cellulaire. Appartenant aux granulomes de type immun, ils sont composés d’une couronne lymphocytaire entourant un follicule central constituée essentiellement de cellules épithéliales associées à des cellules géantes et quelques lymphocytes.
Le poumon sarcoïdien comporte des lésions d’alvéolite, des granulomes et de la fibrose.
– L’alvéolite est la lésion la plus précoce.
– Une fibrose survient en cas d’évolution défavorable.
PATHOGÉNIE ET IMMUNOPATHOLOGIE :
Hypothèses étiologiques :
Parmi les hypothèses étiologiques, on invoque le rôle:
– d’une prédisposition génétique suggérée par certaines études familiales.
– et de la transmission d’agents jusque-là non identifiés appuyée sur des études cas-contact, l’observation d’exceptionnelles micro-épidémies et la survenue d’une sarcoïdose chez deux receveurs d’organes de donneurs atteints.
Immunopathologie :
La sarcoïdose est secondaire à une réponse immunitaire exagérée responsable de la formation de granulomes au niveau des localisations de la maladie.
A ce niveau, les interactions entre monocytes/macrophages et lymphocytes T par le biais de contacts membranaires et la sécrétion de nombreux médiateurs jouent un rôle pathogénique central.
Le profil des cytokines produites par les lymphocytes T est de type TH1. La spécificité antigénique des sous-populations lymphocytaires T n’a pu être précisée, n’offrant actuellement pas de voie de recherche pour une cause.
Atténuation des réactions cutanées tuberculiniques
La sarcoïdose entraîne une atténuation des réactions cutanées tuberculiniques et une lymphopénie, sans induire d’immunodépression patente sur le plan clinique.
Activations des lymphocytes T
* L’hypergammaglobulinémie polyclonale résulte du passage sanguin d’immunoglobulines produites en excès dans les sites de la maladie.
* Au niveau du poumon et des autres organes cibles, on note une accumulation et une activation des macrophages et des lymphocytes.
* Il y a une stimulation des propriétés sécrétoires des macrophages en enzymes (enzyme de conversion de l’angiotensine) et en cytokines IL-1 qui entraîne une activation des lymphocytes T et interféron-gamma.
La majorité des lymphocytes activés l’est de façon indépendante d’antigène.
Formation des granulomes
* Les lymphocytes T auxiliaires expriment des marqueurs membranaires d’activation comme les molécules HLA-DR et le récepteur de haute affinité de l’IL-2 et produisent spontanément de nombreuses lymphokines dont l’IL-2 qui amplifie l’inflammation par son effet mitogène sur les lymphocytes T, mais aussi l’interféron-gamma et un facteur chémotactique des monocytes qui entraînent l’accumulation et la différenciation des macrophages en cellules épithélioïdes et géantes.
* La coactivation réciproque des lymphocytes T et des macrophages conduit à la formation des granulomes.
* Les lymphocytes T sécrètent également des facteurs de croissance et de différenciation des lymphocytes B.
* L’activation prolongée des macrophages participe à la fibrogenèse par la sécrétion de fibronectine, de PDGF et d’IGF1.
Diagnostic :
CIRCONSTANCES DE DÉCOUVERTE :
La sarcoïdose est symptomatique dans plus de la moitié des cas.
Les circonstances de découverte sont, par ordre décroissant:
– découverte fortuite sur une radiographie thoracique.
– signes cliniques respiratoires (toux persistante, dyspnée).
– localisations viscérales autres que respiratoires (surtout ophtalmologiques, cutanées, adénopathies).
– signes généraux (amaigrissement, fièvre et surtout asthénie).
MANIFESTATIONS MEDIASTINO-PULMONAIRES :
Signes cliniques :
– Une toux sèche se rencontre dans un tiers des cas. Une dyspnée est rare au début.
– L’auscultation pulmonaire est normale le plus souvent. Des râles crépitants sont perçus chez 15% des patients avec une atteinte pulmonaire.
– Il n’y a pas d’hippocratisme digital.
– Des signes de coeur pulmonaire chronique sont possibles à un stade tardif.
Signes radiographiques :
La radiographie de thorax a une place importante dans le diagnostic, le pronostic et la surveillance.
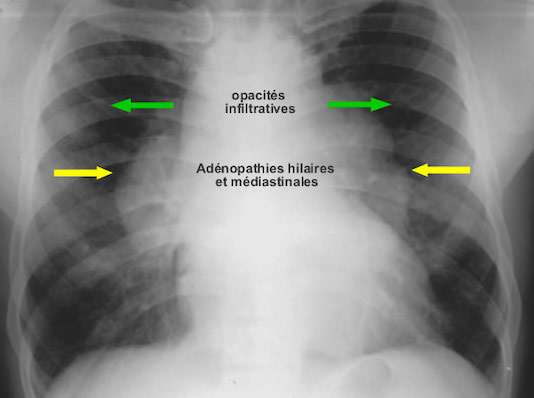
Des signes radiographiques sont observés chez 80 à 90% des patients sous forme d’adénopathies intrathoraciques et/ou d’infiltration pulmonaire diffuse.
De multiples classifications radiographiques ont été proposées, certaines en « stades », d’autres en « types ». Nous avons adopté celle citée ci-après.
Radiographie normale type 0
Adénopathies intrathoraciques isolées type I
* Les adénopathies de type I sont hilaires, bilatérales, symétriques et non compressives.
* Il s’y associe ou non des adénopathies latérotrachéales et/ou sous-carénaires.
* Les adénopathies sont situées préférentiellement dans le médiastin moyen.
* Des calcifications en coquilles d’oeufs peuvent apparaitre après 5 ans d’évolution.
Adénopathies intrathoraciques bilatérales associées à une infiltration pulmonaire ou « réticulo-micronodulaire » type II
* Les micronodules pulmonaires sont diffus dans les champs pulmonaires.
* Rarement, on observe des images en plage arrondies, multiples, pseudo-tumorales d' »allure alvéolaire » ou des opacités en « verre dépoli » diffus.
Infiltration pulmonaire mais sans adénopathies décelables type III
Infiltration pulmonaire diffuse avec fibrose type IV
Opacités linéaires denses associées à une rétraction pulmonaire et à une distorsion trachéo-bronchique.
La rétraction prédomine dans les lobes supérieurs dans la plupart des cas.
Il n’y a pas de corrélation stricte entre la classification radiographique et l’état histopathologique pulmonaire. Cependant, les lésions granulomateuses sont souvent plus importantes aux stades précoces et les lésions de fibrose, aux stades plus évolués.
Signes tomodensitométriques :
La TDM thoracique apporte une contribution diagnostique supplémentaire à la radiographie, particulièrement nette lorsque la radiographie est peu évocatrice.
* Elle permet de repérer des adénopathies infraradiographiques.
* La présence de lésions micronodulaires prédominantes, leur distribution préférentielle péribronchique ou sous-pleurale ainsi que la présence de polygones septaux irréguliers sont suggestives de sarcoidose.
* La TDM permet aussi de mieux déceler certaines complications:
– fibrose pulmonaire.
– greffe aspergillaire.
– bronchectasies.
– compressions (rares) bronchiques ou vasculaires par des adénopathies.
* La TDM permet de distinguer les lésions irréversibles (opacités linéaires, déformation des scissures, distorsions, bronchectasies par traction), a priori de nature fibreuse, de celles potentiellement réversibles sous corticoides (opacités nodulaires ou en plages).
Explorations fonctionnelles respiratoires :
Les anomalies observées ne sont pas spécifiques: syndrome restrictif homogène (fréquent), baisse de la capacité de transfert du CO (très fréquente).
* La courbe débit-volume est souvent anormale. Le rapport VEMS/CV est abaissé chez 5% des patients.
* Les gaz du sang artériels sont normaux au repos sauf dans les formes évoluées.
* Les anomalies des explorations fonctionnelles respiratoires (EFR) et de l’exploration de l’hématose à l’exercice reflètent l’importance globale des lésions pulmonaires. Elles jouent un rôle essentiel dans:
– la décision thérapeutique.
– l’évaluation de la réponse au traitement.
– et la surveillance évolutive.
Endoscopie bronchique et examen cytologique du liquide de lavage broncho-alvéolaire :
Endoscopie bronchique
L’endoscopie bronchique permet la visualisation dans certains cas de granulations endobronchiques voire de lésions pseudo-tumorales et d’épaississements de la muqueuse, deux anomalies associées à une rentabilité biopsique accrue.
– La rentabilité des biopsies de muqueuse bronchique est en moyenne de 60%, à condition d’effectuer au moins trois prélèvements par patient.
– Les biopsies transbronchiques ont une meilleure sensibilité, mais au prix de complications iatrogènes dans 5% des cas et d’une moindre spécificité.
Lavage broncho-alvéolaire
La numération formule cytologique du lavage broncho-alvéolaire reflète la composante luminale de l’alvéolite.
* Il y a typiquement une hypercellularité totale modérée (moins de 500000/ml) avec une augmentation exclusive du pourcentage des lymphocytes et une augmentation du rapport lymphocytes T CD4/CD8.
– Une lymphocytose alvéolaire est mise en évidence dans 80% des cas.
– L’augmentation du rapport lymphocytes T CD4/CD8 est inconstante.
– Une polynucléose neutrophile peut se voir à un stade tardif de la maladie.
* L’intensité de la lymphocytose alvéolaire n’a aucune valeur pronostique.
Scintigraphie au gallium 67 :
La scintigraphie au gallium67 est rarement indiquée. Elle peut montrer une captation anormale pulmonaire, médiastinale ou extra-thoracique (yeux et parotides) parfois suggestive.
Une hypercaptation reflète la présence de lésions granulomateuses actives.
MANIFESTATIONS EXTRA-THORACIQUES :
Les localisations extra-médiastino-pulmonaires sont constatées à un moment ou l’autre de l’évolution dans 20 à 60% des cas selon le recrutement.
Deux fois sur trois, elles sont associées à une atteinte thoracique.
Adénopathies périphériques :
Les adénopathies périphériques sont notées dans 20% des cas.
Elles sont uniques ou multiples et concernent toutes les aires ganglionnaires.
Localisations ophtalmologiques :
Les localisations ophtalmologiques s’observent dans 25% des cas.
La manifestation la plus fréquente est le syndrome sec.
Une uvéite peut être antérieure, intermédiaire ou postérieure.
On peut noter une intumescence des glandes lacrymales.
Une névrite optique est rare.
Manifestations cutanées :
Les manifestations cutanées se voient chez 20% des patients.
* Les sarcoides à petits ou gros nodules, les plaques, les nodules sur cicatrice, le lupus pernio et les nodules sous-cutanés sont des lésions granulomateuses.
* L’érythème noueux n’est pas une lésion sarcoïdienne mais une association morbide privilégiée.
* Le syndrome de Löfgren est l’association d’adénopathies intrathoraciques hilaires bilatérales, d’un érythème noueux et d’une intradermoréaction à la tuberculine négative. Cette dernière condition est en fait facultative. Le pronostic du syndrome de Löfgren est excellent avec une résolution spontanée en moins de 24 mois chez 90% des patients.
Localisations cardiaques :
Les localisations cardiaques spécifiques touchent surtout le ventricule gauche.
Elles donnent des signes caractérisés dans moins de 5% des cas:
* troubles acquis de la conduction auriculo-ventriculaire ou intraventriculaire.
– troubles du rythme ventriculaire.
– douleurs thoraciques parfois angineuses.
– insuffisance ventriculaire gauche progressive.
– troubles de la repolarisation.
– aspect électrocardiographique d’infarctus myocardique.
– péricardite.
– mort subite.
* La tomoscintigraphie myocardique de perfusion au thallium201 ou au MIBI marqué au technétium est très utile au diagnostic.
* L’échographie cardiaque peut montrer diverses anomalies notamment au niveau du système intraventriculaire.
* Il est parfois nécessaire de recourir à une biopsie endomyocardique.
Manifestations neurologiques et musculaires :
Des localisations neurologiques sont reconnues dans 5% des cas.
Elles touchent par ordre décroissant les méninges, le système nerveux central (SNC), les nerfs craniens, les nerfs rachidiens et les muscles. Elles se combinent volontiers entre elles. L’atteinte du VII est très fréquente. Une méningite asymptomatique, lymphocytaire stérile est fréquente.
Les atteintes du SNC sont variées: épilepsie, troubles mentaux ou psychiques, hydrocéphalie, déficits focalisés pseudo-tumoraux, manifestations neuro-endrocrines hypothalamo-hypophysaires (en particulier diabète insipide).
L’IRM cérébrale et médullaire montre des lésions non spécifiques utiles cependant au diagnostic dans le contexte habituel d’une sarcoïdose systémique.
Atteintes rénales :
Les néphropathies interstitielles granulomateuses spécifiques sont rares et leur pronostic réservé.
Les anomalies du métabolisme calcique, une néphrocalcinose ou des lithiases urinaires peuvent entraîner une insuffisance rénale.
Atteintes hépato-spléniques :
* L’atteinte hépatique est très fréquente à l’échelle microscopique.
– Elle occasionne des anomalies biologiques mineures (élévation modérée des phosphatases alcalines et/ou des transaminases) dans 20% des cas.
– La cholestase intrahépatique chronique sarcoïdienne, simulant le tableau d’une cirrhose biliaire primitive (mais sans anticorps antimitochondries) et l’hypertension portale représentent des complications peu fréquentes mais graves.
* Une splénomégalie, avec ou sans hypersplénisme, indique une localisation spécifique ou une hypertension portale.
Parotidite :
* Une parotidite bilatérale indolore est révélatrice dans 5% des cas de sarcoïdose.
* Le syndrome d’Heerfordt associe uvéite, parotidite et fièvre et le plus souvent une atteinte du nerf facial.
Autres localisations :
Tous les autres organes peuvent être touchés avec une fréquence moindre:
– lésions osseuses radiographiques des mains, des pieds (géodes à l’emporte-pièce, aspect grillagé, rupture corticale) ou de la voûte du crâne, arthrites chroniques.
– adénopathies abdominales (à l’échographie ou au scanner abdominal).
– localisations ORL, thyroïdienne, gastriques, épididymite, etc.
SIGNES GENERAUX :
Une fièvre au long cours, des sueurs nocturnes et un amaigrissement parfois marqué accompagnent les formes polyviscérales et évolutives de la maladie, le syndrome de Löfgren, le syndrome d’Heerfordt et parfois les localisations hépatiques ou les adénopathies abdominales. Une asthénie profonde est fréquente.
SIGNES BIOLOGIQUES :
Réactions cutanées tuberculiniques :
La sarcoïdose induit une négativation ou une atténuation des réactions cutanées tuberculiniques dans 80% des cas.
L’anergie n’est que relative et disparaît à la guérison ou en cas de tuberculose.
Recherche d’un agent infectieux :
Les recherches traditionnelles de bacilles tuberculeux et de tout agent infectieux incriminable devant une granulomatose sont négatives.
La sérologie VIH est négative sauf rare coïncidence.
Explorations biologiques courantes :
* La NFS est habituellement normale.
– Une lymphopénie est constatée lorsque la maladie est ancienne et/ou multiviscérale.
– Un syndrome inflammatoire se voit à la phase initiale du syndrome de Löfgren, lors des formes fébriles ou très évolutives. Une anémie hémolytique ou une thrombopénie auto-immunes sont rares.
– Un hypersplénisme peut entrainer une thrombopénie.
* Le bilan hépato-cellulaire est anormal dans 20% des cas.
* Il existe une hypergammaglobulinémie polyclonale dans 50% des cas.
* La fonction rénale est généralement normale.
* Le taux des enzymes musculaires peut être élevé lors des rares atteintes musculaires patentes.
* Les anomalies du métabolisme calcique dues à une hypersécrétion non freinable en calcitriol consistent en une hypercalciurie (dans 40 à 60% des cas) et une hypercalcémie (10% des cas) sans hypophosphorémie. La PTH sérique est basse.
* Le dosage sérique de l’enzyme de conversion de l’angiotensine (ECA) est élevé dans 60% des cas de sarcoïdose, plus souvent dans les formes disséminées à expression intrathoracique et extra-thoracique que dans les formes purement intrathoraciques ou surtout extra-thoraciques pures.
– Il est également plus élevé dans les formes évolutives.
– L’élévation de l’enzyme de conversion de l’angiotensine n’est pas spécifique et peut s’observer lors d’affections granulomateuses ou non, à expression endothoracique ou extra-thoracique.
– L’enzyme de conversion de l’angiotensine n’a aucune valeur pronostique.
– Elle suit fidèlement l’évolution spontanée ou sous traitement de la maladie.
– Le dosage de l’enzyme de conversion de l’angiotensine est particulièrement utile dans l’ajustement du traitement.
PRELEVEMENTS HISTOPATHOLOGIQUES :
Les prélèvements histopathologiques servent à rechercher des granulomes tuberculoïdes sans nécrose caséeuse.
Le choix du prélèvement à effectuer dépend de sa rentabilité a priori, de la plus ou moins grande spécificité d’un résultat positif, de son accessibilité et des risques qu’il entraîne.
– On privilégie les prélèvements perendoscopiques bronchiques et de lésions superficielles (localisations cutanées, adénopathies périphériques).
– Les biopsies de principe de glandes labiales accessoires sont relativement rentables.
– La médiastinoscopie est indiquée devant des adénopathies intrathoraciques visualisées en imagerie et la négativité des recherches plus courantes.
– En l’absence d’adénopathies on propose une biopsie pulmonaire sous vidéothoracoscopie chirurgicale.
– La biopsie hépatique est sensible mais très peu spécifique.
CRITERES DIAGNOSTIQUES :
Compte tenu des inconnues sur l’origine de la maladie et l’extrême variété des présentations rencontrées, le diagnostic de sarcoïdose repose sur trois arguments:
* une présentation radiologique, clinique et biologique évocatrice ou compatible.
* la mise en évidence de lésions granulomateuses sans nécrose caséeuse.
* l’exclusion de toute éventualité d’une autre maladie granulomateuse notamment liée à:
– une infection (tuberculose, histoplasmose, etc.).
– une exposition à des agents minéraux (bérylliose), organiques (pneumopathies d’hypersensibilité) ou à des médicaments.
Il n’est pas nécessaire d’obtenir une confirmation histopathologique dans certaines conditions, comme le syndrome de Löfgren, lorsque la présentation est suffisamment caractéristique.
Il est par contre nécessaire d’obtenir une confirmation histopathologique lorsque la maladie n’est pas typique et/ou qu’un traitement est nécessaire. Il faut souligner ici que la mise en évidence de granulomes n’est pas suffisante à elle seule au diagnostic.
DIAGNOSTICS DIFFERENTIELS :
Les principaux diagnostics différentiels devant une suspicion de sarcoïdose sont:
* devant des adénopathies intrathoraciques:
– lymphome.
– tuberculose.
– métastases.
* devant une atteinte pulmonaire: les autres maladies interstitielles diffuses mais plus particulièrement la tuberculose, la silicose et la bérylliose.
* devant une atteinte extra-thoracique granulomateuse:
– une infection.
– une réaction à corps étranger.
– une origine médicamenteuse.
– une pathologie tumorale de voisinage (lymphome ou cancer).
– une autre granulomatose idiopathique (maladie de Crohn, cirrhose biliaire primitive, granulomatose d’organe).
Devenir de la maladie :
PRONOSTIC :
La probabilité de guérison à court terme est forte durant les deux premières années d’évolution et s’amenuise ensuite.
* Le risque de fibrose pulmonaire est quasi nul dans les deux premières années puis s’accroît pour devenir important au-delà de 5 ans.
* Le pronostic est péjoratif lorsque la maladie débute après 40 ans.
* Les probabilités de guérison spontanée varient selon le type radiographique: type 1 (80%), type 2 (60%) et type 3 (20 à 30%).
* Le risque de complications et de décès s’accroît en sens opposé.
* Un syndrome obstructif (baisse bu VEMS/CV) est péjoratif.
* L’incidence des complications est plus forte chez les patients de couleur.
D’autres signes ont également une certaine valeur pronostique.
* Certains sont favorables:
– latence clinique.
– association d’un érythème noueux.
* les autres sont plutôt péjoratifs:
– dissémination initiale de la maladie.
– évolutivité clinique.
– présence de certaines localisations (SNC, coeur).
– fibrose pulmonaire.
– présence de certains signes (sarcoïdes à gros nodules, lupus pernio, localisations osseuses et ORL).
SURVEILLANCE :
Tous les cas de sarcoïdose doivent bénéficier d’une surveillance périodique jusqu’à la guérison.
Cette surveillance a pour objet la recherche de complications nécessitant une corticothérapie.
* Elle comprend au minimum un examen clinique et une radiographie de thorax tous les 3, 6 ou 12 mois selon l’ancienneté et l’évolutivité de la maladie.
* Une EFR incluant une mesure de la capacité de transfert du CO, un examen ophtalmologique, un ECG et un bilan biologique incluant une NFS, un compte plaquettaire, une étude des fonctions hépatique et rénale ainsi que du métabolisme calcique doivent être contrôlés au moins tous les ans.
* Le dosage de l’enzyme de conversion de l’angiotensine sérique est utile dans les formes sévères et traitées.
ÉVOLUTION SIMPLE :
L’évolution de la sarcoïdose est spontanément favorable dans 50 à 80% des cas: sarcoïdoses asymptomatiques chez des patients blancs ayant une atteinte de « type 1 ou 2 » à la radiographie, sans retentissement aux EFR.
La guérison spontanée survient le plus souvent en moins de 2 ans.
ÉVOLUTION COMPLIQUEE :
Complications respiratoires précoces :
Insuffisance respiratoire subaigue due à une profusion inhabituelle de lésions micronodulaires pulmonaires
La symptomatologie d’une insuffisance respiratoire subaigue due à une profusion inhabituelle de lésions micronodulaires pulmonaires est dominée par une dyspnée progressive sur quelques semaines. Il y a un syndrome restrictif franc et un effondrement de capacité de transfert du CO.
– Il existe une hypoxémie artérielle au repos, aggravée à l’exercice associé.
– Les index d’activité de la maladie sont très pathologiques.
La corticothérapie est remarquablement efficace.
Atteinte sévère des voies aériennes
Les signes révélateurs d’une atteinte sévère des voies aériennes comprennent une dyspnée subaigue, une toux chronique, des sibilances et parfois une hémoptysie. Le diagnostic d’asthme est souvent porté à tort.
La radiographie de thorax est plus ou moins typique.
Au cours de l’endoscopie bronchique, on note une infiltration muqueuse plus ou moins diffuse, un aspect « granité » ou des bourgeons d’allure tumorale. L’EFR met en évidence un syndrome obstructif.
Complications respiratoires tardives :
Les complications respiratoires tardives résultent, au moins en partie, de lésions de fibrose.
– Les lésions de fibrose se développent dans l’interstitium pulmonaire, les bronches proximales ou distales ou bien les vaisseaux pulmonaires.
– Elles sont purement séquellaires d’une maladie passée ou associées à des lésions inflammatoires évolutives (formes actives agressives).
Les quatre principales complications respiratoires tardives sont les suivantes:
* insuffisance respiratoire par fibrose pulmonaire:
– c’est la principale cause d’insuffisance respiratoire.
– elle est souvent évidente sur la radiographie (type IV) et particulièrement bien visible en TDM.
– il existe un syndrome restrictif homogène, une baisse importante de la capacité de transfert du CO, des troubles de l’hématose à l’exercice voire au repos.
– il convient de rechercher des lésions granulomateuses associées par TDM, scintigraphie au gallium67 et lavage broncho-alvéolaire.
* l’aspergillome intracavitaire:
– est révélé par une hémoptysie ou une image radiographique ou tomodensitométrique en grelot.
– la sérologie aspergillaire est positive.
* La bronchopathie chronique obstructive:
– représente la séquelle d’une sarcoïdose bronchique sévère.
– emprunte la présentation clinique et fonctionnelle d’une broncho-pneumopathie chronique obstructive.
– comporte parfois une dilatation des bronches.
* Le coeur pulmonaire chronique résulte à la fois de lésions vasculaires pulmonaires spécifiques et de l’insuffisance respiratoire provoquée par une fibrose pulmonaire.
Complications extra-respiratoires :
(Voir supra « Manifestations extra-médiastino-pulmonaires ».)
Hypercalcémie :
Une hypercalcémie survient dans moins de 10% des cas.
Elle se voit lors des formes polyviscérales et évolutives.
Elle est parfois déclenchée par la prise de vitamine D, une exposition solaire, une consommation excessive de laitages.
Elle est rarement très sévère. Une hypercalcémie peut se voir à un stade précoce mais aussi à distance du début de la maladie.
Corticorésistance :
Une corticorésistance des lésions pulmonaires granulomateuses est exceptionnelle.
Une corticorésistance relative est possible:
– on observe une absence de réponse pour les doses usuellement prescrites (0,5mg/kg/24h).
– mais une réponse complète pour des doses supérieures (1mg/kg/24h).
D’autres localisations de la maladie peuvent être corticorésistantes:
– lésions cutanées (lupus pernio, sarcoïdes à gros nodules).
– cholestase intrahépatique chronique sévère.
– localisations cardiaques, neurologiques, musculaires ou ORL
MORTALITE :
Près de 5% des patients atteints de sarcoïdose en meurent.
Les causes de décès les plus fréquentes sont l’insuffisance respiratoire, le coeur pulmonaire et les hémoptysies de grande abondance par aspergillome, mais aussi les localisations cardiaques, l’atteinte du SNC.
Traitement :
ABSTENTION THÉRAPEUTIQUE :
La moitié au moins des patients guérissent spontanément sans séquelles.
– Les lésions granulomateuses pulmonaires n’entraînent le plus souvent que peu de retentissement fonctionnel.
– Leur résolution précoce en moins de 2 ans empêche l’apparition d’une fibrose.
Les patients asymptomatiques dont la radiographie est classée en type I ou II représentent un gros contingent de malades à ne pas traiter d’emblée.
Devant un syndrome de Löfgren, on préfère se limiter à un traitement de quelques semaines par de la colchicine ou des anti-inflammatoires non stéroïdiens.
INDICATIONS DE LA CORTICOTHERAPIE PAR VOIE GENERALE :
Le bénéfice du traitement corticostéroïde doit être supérieur à ses inconvénients.
Maladie « récente » et mal tolérée :
* Une atteinte pulmonaire entraînant une dyspnée avec anomalies franches de la fonction respiratoire et/ou de l’hématose à l’exercice et des index biologiques d' »activité » anormaux est justifiable d’une corticothérapie. Une granulomatose bronchique avec syndrome obstructif et/ou lésions pseudo-tumorales à l’endoscopie nécessite également une corticothérapie.
* Certaines localisations extra-respiratoires impliquent aussi une corticothérapie: atteintes ophtalmologique non accessible à un traitement local, cardiaque, neurologique centrale, hypothalamo-hypophysaire, rénale, hépatique grave avec cholestase sévère, ORL ou parotidite anormalement durable.
* L’hypercalcémie nécessite le plus souvent une corticothérapie.
Maladie d’évolution « prolongée » :
Un traitement corticostéroïde peut être motivé par deux raisons:
* la première consiste en une nouvelle localisation ou l’aggravation récente d’une localisation antérieurement connue liée à une poussée évolutive granulomateuse, situation proche du cas des « maladies récentes et mal tolérées ».
* la deuxième consiste en la coexistence de lésions granulomateuses associées chez des patients ayant déjà une fibrose pulmonaire avérée:
– les patients traités dans ce cas ont une sarcoidose chronique active fibrosante.
– les investigations complémentaires sont très utiles pour les détecter et adapter le traitement.
– la combinaison d’une dégradation de la fonction respiratoire et de l’hématose à l’exercice et de signes biologiques d’activité représente pour tous une indication thérapeutique.
– même lorsqu’une activité résiduelle n’est pas évidente, une tentative thérapeutique peut être entreprise sur une durée limitée (3 mois).
CONDUITE DE LA CORTICOTHERAPIE :
Traitement d’attaque
le traitement d’attaque est fondé sur la prescription de prednisone ou de prednisolone à raison de 0,5mg/kg/24h pendant 3 mois.
– La posologie doit être plus élevée le premier mois (1mg/kg/24h) en présence de localisations neurologiques centrales, cardiaques, ophtalmologiques sévères, ORL ou d’un échec de la posologie habituelle.
– Une « rémission » des signes potentiellement réversibles est obtenue dans la plupart des cas.
Traitement de décroissance
Le traitement de décroissance est prescrit durant au moins 15 mois avec des doses lentement dégressives par plateaux de 3 mois. Un traitement sur plusieurs années est parfois nécessaire. Il faut dépister un éventuel rebond lors de la décroissance du traitement et dans l’année suivant son arrêt.
Surveillance et précautions d’emploi
* Une surveillance des patients s’impose durant le traitement puis 3, 6 et 12 mois après son arrêt complet. L’absence de rechute 12 mois après l’arret du traitement permet d’affirmer la guérison.
* La corticothérapie nécessite des précautions.
– Le risque de tuberculose sous corticoïdes n’est pas plus important en cas de sarcoïdose que pour d’autres maladies. Chez les patients préalablement exposés à un risque endémique, non vaccinés par le BCG et dont l’intradermoréaction à la tuberculine est positive, une prophylaxie antituberculeuse est recommandée.
– En cas d’ostéoporose cortisonique, la prescription de dérivés de la vitamine D et de calcium est prudente (impliquant un contrôle confirmé de l’activité de la maladie et une surveillance calcique régulière). La calcitonine soulève moins d’interrogations.
ALTERNATIVE AU TRAITEMENT CORTICOSTEROIDE PAR VOIE GENERALE :
En cas de contre-indication ou d’inefficacité des corticoïdes se discute, avec des limites à bien connaître, le recours aux antipaludéens de synthèse ou au méthotréxate.
Traitements locaux par corticostéroïdes
– Les traitements corticoïdes locaux sont utiles en présence de localisations cutanées ou ophtalmologiques.
– Les corticoïdes inhalés ont un effet antitussif appréciable au cours de sarcoïdoses ne relevant par d’une corticothérapie générale et pourraient avoir une place d’épargne en corticoïdes généraux en fin de traitement.
Précautions hygiénodiététiques
Chez les patients non traités:
– la prise de dérivés de la vitamine D parfois inclus dans certaines préparations polyvitaminées est contre-indiquée.
– les apports alimentaires en calcium doivent être rationnés. Les expositions solaires sont déconseillées.
– ces mesures, valables en cas de trouble du métabolisme calcique, sont également recommandables en cas de maladie évolutive même en l’absence d’anomalie métabolique patente.
Traitements synptomatiques
Divers traitements symptomatiques peuvent être utiles: oxygénothérapie, antiarythmiques, pose d’un stimulateur cardiaque, antiépileptiques, voire transplantation d’organe. Les indications de transplantation pulmonaire doivent rester prudentes. La maladie est faiblement évolutive à un stade avancé. La coexistence de localisations extra-respiratoires graves doit être prise en considération.







")



{kind=link}