



















Introduction :
A – DÉFINITION :
L’Organisation mondiale de la santé (OMS) définit l’ostéosarcome comme une « tumeur maligne caractérisée par l’élaboration d’os ou de substance ostéoïde par les cellules tumorales ».
Microscopiquement, cette tumeur mêle différents composants, cartilagineux, fusiforme, ostéoblastique et anaplasique, dont l’importance respective varie d’un patient à l’autre et d’un secteur à l’autre de la tumeur.
 Selon la définition, une production d’os, même minime, suffit pour parler d’ostéosarcome.
Selon la définition, une production d’os, même minime, suffit pour parler d’ostéosarcome.
Celle-ci permet aussi d’écarter les tumeurs avec production d’os réactionnel par le stroma.
L’ostéosarcome comporte une grande variété de lésions distinctes par leur présentation clinique et radiographique, leur aspect microscopique et leur évolution.
Trois groupes sont identifiés selon leur localisation, les ostéosarcomes de surface, ceux de siège intracortical et les ostéosarcomes intramédullaires ou centraux qui sont les plus fréquents.
Les ostéosarcomes intracorticaux sont extrêmement rares et seules des observations isolées sont rapportées.
Les ostéosarcomes développés à la surface de l’os sont généralement de bon pronostic, associant les formes périostées, d’aspect cartilagineux, les formes paraostéales ou juxtacorticales, très ostéoformatrices, et ceux de forme commune, plus agressifs.
Les ostéosarcomes centraux regroupent différentes variantes histologiques qui, à l’exception de la forme dite « bien différenciée intramédullaire », sont toutes de haute malignité.
Il s’agit de l’ostéosarcome télangiectasique, caractérisé par des lacs sanguins, l’ostéosarcome à petites cellules rondes identiques à celles du sarcome d’Ewing et la forme commune scindée, selon le contingent prédominant, en variantes ostéoblastique, chondroblastique ou fibroblastique.
B – TERMINOLOGIE :
Peu de termes ont été utilisés dans le passé : sarcome squelettogène, sarcome ostéogène qui a été le plus usité et progressivement remplacé, dans la nomenclature internationale, par celui d’ostéosarcome.
C – ÉPIDÉMIOLOGIE :
L’ostéosarcome est une tumeur rare, puisque le nombre d’ostéosarcomes découverts par année en France est estimé entre 150 et 200.
Pour comparaison, l’incidence évaluée en Suède, dont le registre des tumeurs est un des mieux tenus, serait de 4,6 cas par million d’habitants.
Malgré ce faible chiffre et si l’on excepte le myélome, l’ostéosarcome est la tumeur osseuse maligne primitive la plus fréquente.
Il représente, dans les grandes séries, environ 20 % des tumeurs malignes et près du double des cas de sarcomes d’Ewing et de chondrosarcomes.
L’incidence serait plus faible dans les populations asiatiques et latinoaméricaines.
Il survient entre 10 et 20 ans et touche entre une fois et demie et deux fois plus fréquemment les garçons que les filles.
Il est rare avant 10 ans, exceptionnel avant 5 ans, quelques cas d’ostéosarcomes congénitaux ont même été décrits.
Très rare aussi après 60 ans où il survient généralement sur terrain prédisposé, l’incidence dans cette tranche d’âge augmenterait pour certains.
D – ÉTIOLOGIE :
Elle est à ce jour inconnue.
Si de nombreuses théories ont été émises à partir de constatations expérimentales ou chez l’animal, comme une origine virale, traumatique, chimique etc, aucune n’a reçu de confirmation chez l’homme. Plusieurs constatations :
– les sujets de grande taille développeraient des ostéosarcomes avec une plus grande fréquence, ce qui est à rapprocher des observations chez les chiens dont certaines races (saint-Bernard, danois) présenteraient plus d’ostéosarcomes ;
– différentes conditions peuvent être associées à l’ostéosarcome, considérées comme des facteurs favorisants ; ces ostéosarcomes, appelés secondaires, surviennent plus tardivement et à des localisations moins habituelles ;
– après irradiation, l’augmentation de son incidence est constamment retrouvée, qu’elle soit militaire, accidentelle, professionnelle ou thérapeutique ; lorsqu’elle est localisée, la tumeur naît généralement en bordure du champ d’irradiation ;
– l’ostéosarcome se développe aussi à partir de tumeurs préexistantes, dysplasie fibreuse, tumeur à cellules géantes, ou bien de maladie osseuse sous-jacente, ostéogenèse imparfaite, mélorhéostose et surtout la maladie de Paget ; il fait suite, plus rarement, à un infarctus ou une ostéomyélite.
Très rarement, l’ostéosarcome peut être familial.
Il survient dans un tableau de tumeurs multiples de différentes variétés histologiques et correspond à la présence d’un gène de prédisposition aux tumeurs.
On peut : citer le syndrome de Li-Fraumeni qui correspond à l’anomalie innée de l’un des allèles du gène p53, l’enfant développant de multiples tumeurs aussi bien sarcomateuses (os et tissus mous) que carcinomateuses ; également le rétinoblastome bilatéral, maladie caractérisée par des tumeurs oculaires bilatérales et où les ostéosarcomes s’observent 100 fois plus fréquemment que dans la population normale.
Les anomalies cytogénétiques observées dans la tumeur sont caractérisées par leur extrême fréquence et leur grande complexité.
Il n’existe pas de lésion spécifique reconnue comme pour le sarcome d’Ewing, mais une fréquente atteinte des locus des gènes p53 et du rétinoblastome.
Localisation :
L’ostéosarcome peut toucher tous les os mais manifeste une prédilection pour la métaphyse des os longs.
Moins de 10 % surviennent à la diaphyse et les localisations épiphysaires sont encore plus rares.
Il se localise surtout au genou : extrémité inférieure du fémur (40 %) et extrémité supérieure du tibia (15 %), puis à l’extrémité supérieure du fémur et de l’humérus (14 %), sites correspondant aux segments osseux dont la croissance est la plus importante de l’organisme.
Les os plats et les os courts sont plus rarement touchés.
Le crâne et la face sont atteints dans moins 10 % des cas, préférentiellement à la voûte et la base du crâne, ainsi que la mandibule.
Ils surviennent à un âge plus avancé et leur forme histologique est plus volontiers chondroblastique.
Localisé au pelvis pour 10 % des cas, il est de grande taille, de variété chondroblastique, et envahit fréquemment les troncs vasculaires.
Il survient secondairement à une irradiation ou une maladie de Paget.
Le rachis représente entre 1 et 3 % des localisations, souvent secondaires.
Il prédomine à la partie inférieure du rachis, naissant du corps vertébral et s’étend rapidement vers le canal médullaire.
Pour tous les os du squelette, même pour les sésamoïdes, des cas d’ostéosarcomes ont été rapportés.
Étude clinique :
La principale manifestation clinique est la douleur au site tumoral, irradiant vers les articulations de voisinage.
Elle débute insidieusement, puis croît, devenant intermittente puis continue, non calmée par le repos ou les antalgiques ordinaires.
Elle est très fréquemment attribuée par le patient à un traumatisme mineur survenant dans les jours précédant la consultation.
Une masse palpable apparaît plus tardivement, sensible à la palpation, pouvant gêner la mobilité de l’articulation selon sa taille.
À un stade plus tardif, des signes inflammatoires cutanés et une stase veineuse sont visibles.
Les fractures pathologiques sont rares, touchant les plus volumineuses lésions.
Le délai entre l’apparition des premiers symptômes et la consultation médicale est plus court que celui du sarcome d’Ewing, en moyenne de 6 semaines, mais le diagnostic n’est porté initialement que dans un tiers des cas.
L’augmentation du taux sérique des phosphatases alcalines est fréquente, conséquence de l’activité ostéoblastique de la tumeur.
Elle n’est pas toujours corrélée au taux mesuré dans la tumeur. Plus caractéristique serait le retour à la normale après exérèse.
La persistance traduirait la présence de métastases ou une excision incomplète.
Étude radiologique :
L’imagerie a évolué rapidement.
À la radiologie conventionnelle, première étape qui reste indispensable, et à la scintigraphie osseuse se sont ajoutés le scanner et l’imagerie par résonance magnétique (IRM).
L’imagerie sert au diagnostic, au bilan local d’extension, au bilan à distance, à l’appréciation de l’efficacité de la chimiothérapie néoadjuvante, à la détection des récidives.
A – DIAGNOSTIC :
La première étape est toujours la radiologie conventionnelle.
S’en passer et aller directement au scanner ou à l’IRM devant des douleurs ou une masse peut conduire à des erreurs grossières.
L’association de paramètres diagnostiques non radiologiques (comme l’âge, la localisation, la taille, la partie de l’os atteinte…) et radiologiques permet une probabilité diagnostique et guide l’attitude pratique.
L’examen repose sur des clichés orthogonaux de tout l’os douloureux, complétés au besoin par des clichés localisés améliorant la résolution spatiale et donc l’étude des petits détails, des incidences obliques pour détecter des anomalies limitées du cortex ou des calcifications des parties molles.
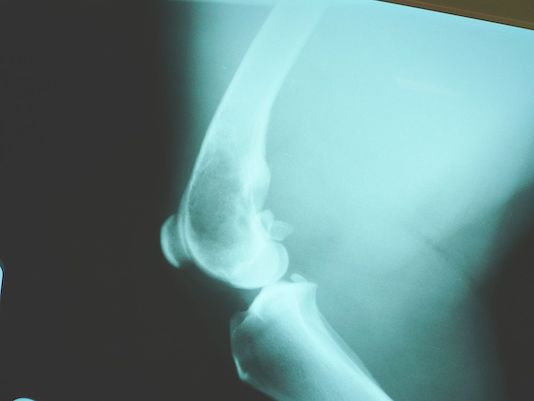
La radiographie révèle le plus souvent des signes de tumeur très agressive.
La tumeur peut être rarement lytique pure, faisant disparaître les travées osseuses, condensante pure, ossifiée ou contenant des calcifications de type cartilagineux (arciformes à centre clair).
Une biopsie dans une région de ce type peut amener un diagnostic de chondrosarcome, soulignant l’intérêt d’une confrontation multidisciplinaire.
Le plus souvent elle est mixte, associant condensation et lyse.
La limite de la tumeur avec l’os normal donne une image de sa rapidité d’évolution.
Celle des ostéosarcomes est le plus souvent de type perméatif, faite de multiples petites lésions lytiques (type 3), la transition entre l’os normal et pathologique ne pouvant être déterminée précisément. Les réactions du périoste à la progression tumorale sont très fréquentes, et là encore traduisent une tumeur agressive.
Les spicules périostés perpendiculaires, souvent divergents (en « coucher de soleil »), sont fréquents et les appositions parallèles sont souvent rompues en leur centre par la rapide progression tumorale (éperon de Codman).
L’envahissement des parties molles, quasi constant, n’est évident que si la tumeur est calcifiée, sinon mieux précisé en scanner ou IRM.
Au total, l’aspect de lésion agressive, souvent très évocateur du diagnostic, doit conduire à compléter l’examen par d’autres techniques d’imagerie, si possible avant la biopsie, qui fausse le bilan d’extension.
Le scanner est demandé s’il existe des problèmes diagnostiques ou si l’IRM n’est pas disponible pour le bilan d’extension.
Il est particulièrement efficace dans l’étude des os courts et plats, rares sièges d’ostéosarcome.
Dans ces localisations difficiles à étudier sur les clichés conventionnels, le scanner est une précieuse aide diagnostique, révélant des lésions lytiques ou condensantes, une atteinte limitée du cortex, des appositions périostées et la lésion des parties molles.
Mais parfois, même pour les os longs, il peut améliorer l’analyse de la lésion. Ainsi, de petites calcifications, de minimes appositions périostées perpendiculaires, un envahissement des parties molles, une atteinte de part et d’autre d’un cortex paraissant encore intact (et traduisant ainsi la rapidité de progression tumorale), peuvent augmenter la suspicion de malignité.
Une bonne technique utilise des coupes d’épaisseur adaptée (fines pour les os longs) et les possibilités de traitement d’image (agrandissements, fenêtrages différents pour l’os et les parties molles, inversion d’image pour analyser l’os dense, mesures de densité).
L’injection de produit de contraste n’est pas utile quand l’IRM est également réalisée, la visualisation des parties molles étant supérieure en IRM.
Celle-ci ne permet pas une bonne analyse des calcifications et donc sa valeur diagnostique est très limitée. Les mesures des temps de relaxation et les études dynamiques après injection n’ont qu’une valeur diagnostique très limitée.
En revanche, le contraste très supérieur et le choix du plan de coupe sans déplacer le patient rendent l’IRM très supérieure dans le bilan d’extension (malgré les nouvelles possibilités du scanner avec reconstructions 3D).
B – FORMES PARTICULIÈRES :
1- Ostéosarcome de haut grade de surface :
Cette tumeur rare est un ostéosarcome conventionnel, dont elle partage les aspects en imagerie, les problèmes thérapeutiques et le pronostic, mais se développe sur le versant externe de la corticale osseuse.
Elle est le plus souvent ossifiée, parfois très dense.
2- Ostéosarcome télangiectasique :
La tumeur est purement lytique.
Des lacs sanguins sont parfois visibles en scanner et surtout en IRM.
Des nodules solides et une paroi épaisse et irrégulière doivent faire suspecter le diagnostic.
C – APPRÉCIATION DE L’EFFICACITÉ DE LA CHIMIOTHÉRAPIE NÉOADJUVANTE :
Il serait utile de prévoir l’efficacité de la chimiothérapie préopératoire avant la chirurgie et l’étude histologique, pour adapter plus tôt le traitement.
Sur les clichés standards et le scanner, une diminution de la masse tumorale, une meilleure limitation et plus d’ossification indiquent une bonne efficacité du traitement.
Les exceptions sont néanmoins trop nombreuses pour permettre un usage pratique.
L’IRM permet une visualisation plus précise : sans injection, la persistance ou l’aggravation de l’oedème péritumoral sur les séquences à pondération T2 indiquent un mauvais répondeur.
Surtout, les séquences après injection et acquisition rapide et répétée permettent de localiser directement le tissu tumoral dans l’image : la prise de contraste précoce (dans les 2 premières minutes) visualise directement le tissu tumoral viable, l’inflammation ayant un rehaussement plus tardif.
Différentes techniques (analyse factorielle des structures dynamiques, soustraction, imagerie de premier passage) peuvent être réalisées, avec des résultats proches.
Elles sont maintenant souvent disponibles en routine sur la console de la machine et fonctionnent très rapidement.
Il est possible de superposer la localisation du tissu tumoral viable à l’image anatomique.
Malheureusement, si cette information est fiable, elle ne le devient que juste avant la chirurgie (on ne peut donc pas prévoir l’efficacité du traitement après seulement une cure de chimiothérapie et modifier le traitement en conséquence).
D – DÉTECTION DES RÉCIDIVES :
Localement, la surveillance après pose d’une prothèse est assurée au mieux en IRM si le patient a une prothèse non paramagnétique en titane, ce qui n’est pas toujours le cas.
Sa place est relative pour les ostéosarcomes des membres où elle est surtout clinique.
La surveillance thoracique est assurée par clichés standards et scanners.
Bilan d’extension :
A – BILAN LOCAL D’EXTENSION :
Il repose sur l’IRM.
Celle-ci comporte des plans orthogonaux, adaptés à la région, avec antenne de surface si possible, améliorant la résolution spatiale, des séquences à pondérations variées (T1 pour analyser la moelle osseuse, T2 ou séquence d’inversion-récupération [STIR] pour les parties molles).
L’injection de produit de contraste n’est pas systématique dans le bilan initial.
L’IRM permet de bien délimiter la tumeur :
– dans la moelle osseuse diaphysaire, permettant de choisir le niveau de la résection ; les skip-metastases (séparées de la tumeur principale par du tissu normal) sont très rares ; elles atteignent le plus souvent le même os, mais parfois un autre proche ;
– vers le cartilage de croissance, qui est très bien vu ; son franchissement est parfaitement analysé ; l’analyse de l’extension dans l’os spongieux métaphysaire et éventuellement épiphysaire est facile, alors qu’elle est très peu performante en scanner ;
– l’atteinte des parties molles, de la peau, est facilement évaluée ;
– l’analyse des vaisseaux (rarement envahis et seulement refoulés d’ordinaire) peut être aussi réalisée en IRM ; il est possible de réaliser une artériographie-IRM aussi performante qu’un examen conventionnel, sans ou avec une injection intraveineuse de peu de contraste ; l’unique indication de l’artériographie conventionnelle reste l’injection intra-artérielle de chimiothérapie.
La principale limite de l’examen est l’impossibilité de préciser si une tumeur qui va jusqu’au cartilage articulaire envahit l’articulation.
Bien sûr les atteintes massives ne posent pas de problèmes.
Cette limite, parfois très gênante, est reconnue par toutes les équipes, et les signes indirects (épanchement articulaire) ne sont pas plus fiables.
L’oedème péritumoral peut parfois poser des problèmes, faisant surévaluer la tumeur, surtout sur les images pondérées T2 et STIR.
Dans les cas difficiles, le contrôle après chimiothérapie permet le plus souvent de répondre.
B – BILAN D’EXTENSION À DISTANCE :
Il repose sur le cliché de thorax et le scanner pour les métastases pulmonaires, la scintigraphie pour les plus rares métastases osseuses.
Le scanner pulmonaire fait partie du bilan systématique, même si sa sensibilité et sa spécificité sont médiocres (nondétection des petits nodules, très nombreux faux positifs).
La chirurgie étant la meilleure, et probablement la seule chance de guérison des patients métastatiques au poumon, le scanner reste l’examen de référence, en attendant une méthode plus performante.
Anatomie pathologique :
A – BIOPSIE :
L’aspect microscopique est dominé par l’hétérogénéité tumorale et par les fréquents remaniements présents.
Le diagnostic est entièrement conditionné par le site de la biopsie chirurgicale et le volume des fragments et nécessite impérativement la transmission au laboratoire des informations cliniques et radiographiques (cliché standard) et du compte rendu opératoire.
En leur absence, le pathologiste n’est pas tenu d’effectuer un diagnostic formel.
La démarche diagnostique du pathologiste est d’abord d’affirmer la malignité de la tumeur, de reconnaître l’ostéoformation par les cellules tumorales et enfin de préciser la forme histologique.
1- Affirmer la malignité de la tumeur :
Plus facile que pour les autres formes d’ostéosarcomes, elle associe généralement une cellularité très importante et des anomalies cytologiques, polymorphisme, inversion du rapport nucléocytoplasmique, mitoses abondantes et fréquemment anormales, qui sont caractéristiques.
Lorsque la lésion est mieux différenciée, les anomalies cytonucléaires peuvent être minimes, voire absentes, et ce n’est que l’examen attentif à la recherche de mitoses anormales et la corrélation aux données cliniques et radiographiques qui permettent d’établir le diagnostic.
2- Reconnaître l’ostéoformation par les cellules tumorales :
L’ostéoformation est facilement reconnaissable, sous forme d’os non lamellaire constituant des travées de taille et d’épaisseur variables, d’agencement anarchique et directement bordé par les ostéoblastes tumoraux.
Parfois très abondante, la substance ostéoïde peut réaliser des structures plus régulières, organoïdes, se moulant autour des lamelles osseuses normales préexistantes.
Sa répartition varie selon le site, plus abondante au centre de la tumeur, dans le fût diaphysaire, plus faible en périphérie où elle est associée à l’ostéogenèse réactionnelle du périoste et de la corticale.
L’ostéoformation peut être parfois minime, disposée en petits îlots ou en fin réseau entourant chaque cellule, difficile à distinguer de fibres de collagène, même par examen en lumière polarisée.
À la différence d’autres types tumoraux, il n’existe aucun immunomarquage spécifique de l’origine ostéoblastique des cellules ni de la nature ostéoïde de la substance élaborée.
Le diagnostic repose uniquement sur l’aspect morphologique et dépend du caractère représentatif de la biopsie.
3- Classer la lésion :
En plus de la forme anatomopathologique, il est généralement demandé au pathologiste de typer la lésion selon l’une des différentes classifications pronostiques existantes.
La plus répandue est le grading de Broders qui comporte quatre degrés différents (I à IV).
Son intérêt apparaît restreint car la majorité des ostéosarcomes sont à classer comme grade IV ou III, sa reproductibilité n’a jamais été établie et semble limitée et sa valeur pronostique, comme pour les autres classifications, n’est pas significative.
B – DIFFÉRENTES FORMES ANATOMOPATHOLOGIQUES ET LEUR SIGNIFICATION ÉVENTUELLE :
1- Forme commune :
De loin la plus fréquente, elle a été classée par Dahlin selon le contingent prédominant.
La variante ostéoblastique représente environ 50 % des cas, les variantes chondroblastique et fibroblastique 25 % chacune.
Cette classification, conçue initialement à des fins pronostiques, s’est progressivement généralisée, mais sa fiabilité apparaît imparfaite, limitée par l’hétérogénéité tumorale et par la représentativité partielle du matériel biopsique dont le volume représente souvent moins de 1 % de la masse tumorale.
2- Télangiectasique :
Sa fréquence varie selon les séries entre 2 et 10 %, différences liées aux critères anatomoradiologiques retenus.
Lésion purement lytique et expansive sur la radiographie, à l’origine de fréquentes fractures pathologiques, son aspect lors de la biopsie et en macroscopie est celui d’une lésion multilobée à contenu hémorragique.
Microscopiquement, les cavités hémorragiques sont séparées par des cloisons fibreuses d’épaisseur variable mêlant des cellules géantes multinucléées, sidérophages, de l’ostéoïde en travées grêles et la population tumorale souvent polymorphe et atypique.
Huvos a intégré à cette forme les ostéosarcomes avec une importante nécrose hémorragique.
Considérée par Dahlin comme la forme d’ostéosarcome la plus agressive, son pronostic est aujourd’hui proche de celui des formes standards.
3- À petites cellules rondes :
D’aspect radiographique hétérogène, la centaine de cas décrits concerne des jeunes patients entre 10 et 25 ans.
La tumeur est constituée de petites cellules tumorales aux noyaux arrondis, au cytoplasme peu abondant et aux limites imprécises, similaire au sarcome d’Ewing.
L’ostéoïde élaboré par les cellules est en quantité très variable selon les cas, accompagné rarement par des plages cartilagineuses.
Le pronostic est péjoratif et certaines équipes les traitent selon les protocoles des sarcomes d’Ewing.
4- Ostéosarcomatose :
Elle correspond à la survenue de plusieurs ostéosarcomes de siège différent, soit de façon simultanée (synchrone), soit différée dans le temps (asynchrone).
Cette entité très particulière s’observe à la fois chez l’adulte et l’enfant.
Il s’agit d’ostéosarcomes intramédullaires développés dans des os longs, de répartition souvent symétrique et de même taille.
Le pronostic est extrêmement péjoratif, avec une moyenne de survie de quelques mois.
C – PIÈCE DE RÉSECTION :
Son étude permet d’apprécier la taille et l’extension de la tumeur, de juger de la qualité de l’exérèse chirurgicale, d’évaluer la réponse à la chimiothérapie et de rechercher une lésion préexistante.
Elle nécessite que le pathologiste dispose de la biopsie initiale pour pouvoir comparer les aspects histologiques avant et après traitement.
Sa prise en charge est bien codifiée.
Avant ouverture de la tumeur, le pathologiste effectue les prélèvements des limites de résection et des masses appendues à la pièce osseuse, muscles, ligaments, capsule articulaire, revêtement cutané etc.
La pièce est radiographiée pour juger de la localisation de la tumeur, puis coupée à la scie selon le plus grand axe de la tumeur en tranches de quelques millimètres d’épaisseur.
La tumeur, toujours hétérogène, associe des zones blanchâtres, charnues ou dures lorsqu’elles sont ossifiées ou calcifiées, des plages bleutées cartilagineuses et des secteurs encéphaloïdes.
Les remaniements sont nombreux et étendus, hémorragiques, kystiques et nécrotiques, modifiant l’aspect, qu’ils soient spontanés ou induits par le traitement ou par une fracture pathologique.
La taille de la tumeur est évaluée sur les tranches de section, en précisant ses deux plus grandes dimensions ainsi que les distances qui la séparent des berges.
La tumeur occupe généralement toute la cavité médullaire, déforme la pièce osseuse, rompt très rapidement la corticale et envahit les tissus mous adjacents.
Sa délimitation est souvent excellente après chimiothérapie.
Elle bute sur le cartilage de croissance qui constitue une barrière anatomique efficace à l’avancée tumorale et à son extension dans l’épiphyse.
De même, l’envahissement intraarticulaire est très rare grâce à la résistance encore plus grande du cartilage articulaire.
Il s’effectue en général par le contingent extraosseux ou aux points de faiblesse, jonction os-cartilage et insertions capsuloligamentaires.
La recherche de skip-metastases est systématique.
Elle correspond à une dissémination osseuse intramédullaire à distance de la tumeur.
Définie par Enneking comme un foyer tumoral isolé de l’ostéosarcome situé dans le même segment osseux mais sans connexion avec la tumeur principale ou bien dans la pièce osseuse opposée (transarticulaire), sa fréquence est très variable suivant les études et les techniques employées mais serait faible, de l’ordre de 5 %.
Microscopiquement, la tumeur après chimiothérapie d’induction est parfois strictement inchangée, mais comporte le plus souvent d’importantes altérations à la fois cellulaires et architecturales.
Elle peut être entièrement stérilisée ou seulement par zone, remplacée par un tissu oedémateux riche en vaisseaux ou par d’épaisses travées osseuses déshabitées et disposées en réseau plus ou moins régulier.
Les cellules tumorales viables sont volumineuses, aux noyaux irréguliers, monstrueux, parfois multiples, aux cytoplasmes vacuolisés.
Elles peuvent être isolées dans un tissu hyalin dense ou disposées en « plages ».
L’ostéoformation est beaucoup plus importante après chimiothérapie.
Son origine est discutée, maturation directement induite par la chimiothérapie, réaction cellulaire passive à une agression.
Les plages résiduelles viables prédominent en des secteurs anatomiques précis, appelés « sites sanctuaires ».
Elles sont situées au contact du périoste, du cartilage articulaire, du cartilage de croissance, le long des vaisseaux et dans l’extension extraosseuse de la tumeur.
Pour apprécier la viabilité après chimiothérapie, le pathologiste sélectionne une tranche entière de section de la tumeur qui est radiographiée, fixée, décalcifiée et incluse en totalité avec réalisation de prélèvements en grille, chacun étant identifié et noté sur un schéma qui accompagne le compte rendu.
Le pathologiste apprécie microscopiquement pour chaque bloc le pourcentage de cellules résiduelles qui est retranscrit sur le schéma puis, après addition des résultats des différents blocs, calcule la moyenne.
Ce protocole établi par Huvos n’est fiable que si le pathologiste dispose de la biopsie avant tout traitement pour juger des modifications survenues.
Le chiffre du pourcentage de cellules résiduelles sert à établir le grade de Rosen, facteur pronostique important et surtout permettant le choix de la chimiothérapie adjuvante. Bien que sa reproductibilité soit régulièrement critiquée, les valeurs obtenues dans les différentes études publiées sont très similaires et sa valeur pronostique pratiquement constante.
Cette valeur pronostique est obtenue au prix d’un nombre considérable de prélèvements pour le laboratoire qui nécessite un temps de technique et de lecture important.
Diagnostic différentiel :
A – RADIOLOGIQUE :
Il se pose surtout avec les autres lésions osseuses agressives, formes initiales d’ostéomyélite (ce qui souligne l’intérêt d’un prélèvement bactériologique systématique lors de la biopsie), rarement granulome à cellules de Langerhans (posant plus de problèmes avec le sarcome d’Ewing), autres tumeurs malignes primitives (Ewing de localisation plus volontiers centrale, souvent fébrile, fibrosarcome, fibrohistiocytome malin, lymphome).
Le sarcome télangiectasique peut simuler un kyste osseux anévrismal devant les lacs sanguins. Des parois épaisses et des nodules pleins en scanner ou IRM doivent attirer l’attention.
B – ANATOMOPATHOLOGIQUE :
En raison de la diversité des aspects histologiques de l’ostéosarcome, le diagnostic différentiel comporte un large éventail de lésions, bien que la forme de haut grade soulève moins de difficultés diagnostiques que les formes bien différenciées.
Malgré tout, la corrélation avec les images radiographiques et les informations cliniques est toujours indispensable, base essentielle du diagnostic en pathologie osseuse et, en son absence, le pathologiste peut ne pas porter de diagnostic de certitude.
1- Variante ostéoblastique :
Bien que la malignité soit souvent patente, deux affections peuvent simuler parfaitement l’ostéosarcome ostéoblastique et doivent donc être systématiquement écartées avant de porter ce diagnostic : ce sont le cal fracturaire et l’ostéoblastome.
* Cal fracturaire :
À un stade précoce, la réaction induite par la fracture est constituée de plages denses d’ostéoblastes immatures avec de nombreuses mitoses, élaborant une substance ostéoïde grêle à disposition anarchique, l’ensemble mimant exactement un ostéosarcome.
En l’absence de notion de traumatisme ou d’évolution rapide de la lésion, le pathologiste peut effectuer à tort un diagnostic de malignité sur les seuls critères morphologiques.
Ceci explique la nécessité absolue pour le pathologiste de disposer des données cliniques, d’une radiographie de la lésion et du compte rendu opératoire pour limiter le risque de faux positifs auquel tout pathologiste a été ou est un jour confronté.
À un stade plus tardif, le diagnostic histologique aussi bien que radiologique devient plus facile, la lésion apparaît moins cellulaire et perd sa disposition anarchique pour s’organiser dans l’espace.
En cas de doute diagnostique, un délai d’observation d’une dizaine de jours sans aucun traitement permet d’apprécier l’évolution spontanée de la lésion.
C’est un vrai problème qui se pose de façon aiguë chez un adolescent dans les suites d’arrachements apophysaires (épine iliaque antéro-inférieure, tubérosité ischiatique).
Le diagnostic est tout aussi difficile dans certaines pathologies d’insertion chez l’enfant sportif (tendons du grand fessier, du grand adducteur sur le fémur, tendon rotulien sur la tubérosité tibiale) pouvant simuler cliniquement, radiologiquement et microscopiquement un ostéosarcome.
* Ostéoblastome :
Développé entre 20 et 30 ans, il peut être de grande taille et agressif, soufflant ou détruisant en partie la pièce osseuse.
Microscopiquement, la distinction est aussi difficile car l’ossification est irrégulièrement répartie et la lésion est cellulaire avec parfois quelques atypies cytonucléaires.
Mais le polymorphisme cellulaire est moins marqué que celui de l’ostéosarcome, la nécrose est absente et les mitoses rares, sans image anormale.
Il faut rappeler que certains ostéosarcomes comportent peu ou pas d’anomalies cytonucléaires, ce qui rend la confrontation radiohistoclinique essentielle.
2- Variante chondroblastique :
Le problème diagnostique se pose avec le chondrosarcome, soit parce que la biopsie n’a pas intéressé les secteurs ossifiés soit parce qu’ils sont très peu abondants (os plats) ou difficiles à reconnaître.
L’âge, la localisation et l’aspect radiographique en complément de l’histologie permettent le diagnostic.
3- Variante fibroblastique :
Le problème est identique pour la composante fusiforme, si ce n’est qu’elle peut être prise pour un fibrosarcome, un histiocytofibrosarcome et un léiomyosarcome, plus rarement un carcinome sarcomatoïde.
L’immunohistochimie apporte une aide indirecte en montrant que la tumeur ne comporte aucune de ces différenciations, épithéliale, musculaire etc.
4- Ostéosarcome télangiectasique :
En microscopie, il s’agit, comme en radiologie, de le différencier du kyste osseux anévrismal.
La présence d’anomalies cytonucléaires permet de trancher en faveur de l’ostéosarcome bien qu’elles soient parfois peu abondantes et nécessitent un examen de l’intégralité des fragments.
5- Variante à petites cellules rondes :
Elle pose le problème de sa distinction avec le sarcome d’Ewing, les autres tumeurs comme un lymphome, le carcinome à petites cellules ou le neuroblastome étant facilement écartées par l’étude immunohistochimique.
La rareté des cas décrits ne facilite pas la recherche de facteurs discriminants.
La cytologie est identique dans les deux affections et l’expression immunohistochimique du mic2 peut même s’observer dans l’ostéosarcome.
C’est dans ce cas que la cytogénétique et la biologie moléculaire se révèlent utiles au diagnostic.
6- Formes de surface :
Pour ces formes, le danger est de méconnaître le contingent de haut grade qui conditionne le traitement chimiothérapique par rapport aux formes périostée et juxtacorticale dont le traitement est surtout chirurgical.
7- Ostéosarcomatose :
Cette circonstance rare doit faire éliminer une dissémination osseuse d’une seule tumeur qui est généralement plus volumineuse et, chez l’adulte, des métastases d’un carcinome induisant une forte production osseuse réactionnelle.
Évolution et pronostic :
A – ÉVOLUTION :
L’évolution spontanée s’effectue d’abord localement, la tumeur devenant volumineuse, envahissant les tissus mous, l’articulation, comprimant les axes vasculonerveux, se compliquant de fractures pathologiques, ce qui rend progressivement le membre non fonctionnel.
La dissémination métastatique survient aussi rapidement.
Lorsque l’amputation était le seul traitement de l’ostéosarcome, environ 80 % des enfants développaient des métastases viscérales dans l’année qui suivait la chirurgie.
La voie hématogène constitue le principal mode de dissémination, avec une prédilection pour les poumons, parfois d’autres pièces osseuses ou la plèvre.
Les métastases ganglionnaires sont inhabituelles, observées entre 1 et 10 % selon les séries.
Depuis la chimiothérapie dont l’avènement a transformé le pronostic, les disséminations métastatiques ne s’observent que dans 20 à 30 % des cas, toujours pulmonaires mais aussi osseuses, de survenue toutefois plus tardive et en nombre plus réduit qu’en l’absence de traitement.
B – PRONOSTIC :
Il doit être considéré sur le plan local, puis général.
Les rechutes locales surviennent avec une même fréquence après amputation ou traitement conservateur, entre 2 et 5 %.
Elles sont liées avant tout à la qualité d’exérèse et sont le prélude à une dissémination générale.
L’évolution est alors extrêmement rapide.
Le pronostic général est régi par deux paramètres essentiels :
– la réponse à la chimiothérapie établie sur la pièce de résection ;
– la présence initiale ou la survenue de métastases ; leur découverte fait passer le pronostic d’environ 70 % à seulement 30 % ; de cette catégorie au pronostic sombre, le délai de survenue des métastases et leur nombre viennent en moduler l’évolution, plus sévère si la dissémination est précoce et multiple, meilleure si elle survient après plusieurs années de surveillance et limitée à quelques nodules ; la réponse à la chimiothérapie apparaît souvent identique à celle de la tumeur principale lorsqu’elles sont concomitantes.
D’autres facteurs comme la taille (> 10 cm) et le volume tumoral (> 150 cm3), le respect ou non du cartilage de croissance chez les enfants, le siège, le caractère bien ou non différencié de l’ostéosarcome, la présence d’une nécrose spontanée, l’augmentation des phosphatases alcalines et des lacticodéshydrogénases (LDH) sériques sont fréquemment retrouvés, mais leur valeur pronostique s’avère largement inférieure à celle des deux premiers paramètres.
De nouveaux paramètres biologiques, comme la p53, la multidrug resistance (MDR), le gène du rétinoblastome etc, font l’objet de nombreuses études et suscitent d’importants espoirs.
Orientation thérapeutique :
A – INTRODUCTION :
Comme toute la démarche diagnostique, le traitement actuel de l’ostéosarcome est multidisciplinaire.
Cela impose une prise en charge par une équipe spécialisée et rodée au traitement de ce type de tumeur, et apte à prendre en charge les éventuelles complications.
La combinaison chimiothérapie d’induction, chirurgie et chimiothérapie adjuvante a transformé le pronostic de cette lésion auparavant gravissime.
La radiothérapie n’est que rarement utilisée du fait de la radiorésistance de ce type de tumeur.
B – TRAITEMENT ADJUVANT :
1- Formes localisées des membres :
Le rôle de la chimiothérapie en complément du geste chirurgical pour le traitement des ostéosarcomes localisés des membres a été bien établi.
Deux études randomisées comparant un groupe de patients traités sans chimiothérapie et un groupe de patients traités avec chimiothérapie adjuvante ont mis en évidence une différence de survie en faveur du groupe de patients traités par chimiothérapie.
Même s’il n’existe pas de preuves formelles de l’avantage d’effectuer une chimiothérapie première par rapport à une chimiothérapie adjuvante sur la survie globale, la diminution significative du taux d’amputation et l’importance de la valeur pronostique de la réponse histologique expliquent l’intérêt de cette séquence thérapeutique désormais standardisée : chimiothérapie d’induction suivie du geste chirurgical et d’une chimiothérapie adjuvante dont la nature dépend du pourcentage moyen de cellules viables sur la pièce opératoire.
On considère qu’un patient est bon répondeur s’il persiste moins de 5 % de cellules viables (grade IV, aucune cellule viable ; grade III, moins de 5 % de cellules viables ou quelques cellules disséminées sur toute la tranche de section) selon la classification histopronostique universellement reconnue établie par Huvos en 1977.
Si la chirurgie après chimiothérapie demeure indispensable, le protocole optimal (produits et durée) de la chimiothérapie des ostéosarcomes reste à déterminer.
Quatre antimitotiques sont actifs (plus de 20 % de réponse en monothérapie) dans cette maladie : le méthotrexate à hautes doses (MTX), la doxorubicine (adriamycine [A]), le cisplatine (CDDP [P]) et l’ifosfamide (I). Les protocoles de chimiothérapie les plus utilisés depuis le début des années 1970 (T7, T10, T12, SFOP [Société française d’oncologie pédiatrique], COSS) comportent tous du MTX à hautes doses associé à de l’acide folinique qui demeure encore, à l’aube du troisième millénaire, la drogue de référence dans les ostéosarcomes localisés, tout au moins dans la population pédiatrique.
Plusieurs enseignements découlent des études pionnières contenant du MTX : le MTX ne doit plus être utilisé seul, mais en association avec l’un ou plusieurs autres produits précités, notamment la doxorubicine ; l’administration de certaines drogues par voie intra-artérielle, notamment le cisplatine, n’augmente pas le taux de réponse histologique par rapport à la voie veineuse ; les fortes doses de MTX doivent être privilégiées au détriment des doses modérées moins efficaces ; le métabolisme, et donc la toxicité, du MTX à hautes doses varie considérablement de l’enfant à l’adulte. Si le MTX doit être administré à la dose d’au moins 12 g/m2 chez l’enfant, il ne faut pas dépasser la dose de 8 g/m2 chez l’adulte.
La méthotrexatémie serait un facteur pronostique important sur la réponse et la survie dans certaines études.
Enfin, la chimiothérapie postopératoire des patients présentant une mauvaise réponse histologique doit être différente de celle utilisée lors de la chimiothérapie d’induction.
Il est remarquable de constater la corrélation linéaire entre la réponse histologique et la survie globale dans toutes les études comportant un nombre conséquent de patients et méthodologiquement bien menées.
Les facteurs pronostiques initiaux comme la taille tumorale, le sous-type histologique, l’âge du patient, la localisation tumorale, le taux initial des LDH ou des phosphatases alcalines sont éclipsés par le paramètre « réponse histologique » lors de l’analyse fine de la pièce opératoire.
Il est relativement aisé de rapporter le pourcentage de bons répondeurs à un taux de survie à 5 ans pour l’ensemble des patients inclus dans une étude donnée.
Ainsi, un taux de bons répondeurs de 30, 40 et 50 % se traduit à 5 ans par une survie globale d’environ 50, 60 et 70 %.
La tolérance médiocre du MTX chez les patients âgés de plus de 18 ans nécessitant fréquemment l’interruption prématurée de la chimiothérapie d’induction, la longueur et la lourdeur logistique (administration hebdomadaire, surveillance rigoureuse et hospitalisations répétées) des schémas thérapeutiques comportant du MTX sont à l’origine de protocoles thérapeutiques dépourvus de ce produit.
Récemment, il a été rapporté dans une large étude multicentrique de l’European Osteosarcoma Intergroup (EOI) rassemblant plus de 400 patients, que le protocole associant la doxorubicine et le cisplatine (AP) donnait des résultats équivalents à une chimiothérapie dérivée du T10 dans les ostéosarcomes opérables, en termes de réponse histologique, survie sans récidive et survie globale.
Bien que l’on connaisse la discordance entre les résultats rapportés des phases II monocentriques et des études de phase III multicentriques, le taux de bons répondeurs (30 %) observés dans le bras thérapeutique contenant du MTX est très éloigné de celui, 50 à 70 %, observé dans les études pilotes.
Dans une pathologie curatrice qu’est l’ostéosarcome localisé, il est aujourd’hui difficile d’accepter un bras thérapeutique de référence (AP) qui ne « guérit » qu’un patient sur deux à 5 ans.
Des approches thérapeutiques similaires sont cependant poursuivies chez l’adulte jeune avec des protocoles intensifiés toujours sans MTX.
Une étude de phase II a ainsi été menée à l’Institut Gustave Roussy (IGR) de 1992 à 1998.
La chimiothérapie d’induction associait doxorubicine-ifosfamide-cisplatine (API) à j1 et j28 et doxorubicineifosfamide (AI) à j15 et j43.
Chaque cure de chimiothérapie (API et AI) était systématiquement suivie de G-CSF afin d’instaurer un intervalle de 15 jours entre chaque cure et de maintenir ainsi une dose-intensité optimale de chaque drogue.
Cette chimiothérapie d’induction se caractérisait par un court délai entre la première cure et l’exérèse chirurgicale (60 jours) et par l’incorporation précoce de l’ifosfamide.
Les patients bons répondeurs recevaient deux cures d’API en situation adjuvante toutes les trois semaines et les mauvais répondeurs trois cures de chimiothérapie associant étoposide et ifosfamide à fortes doses toutes les 4 semaines. Vingt-six patients ont été inclus dans cette étude prospective.
La toxicité de ce protocole était exclusivement hématologique avec des épisodes de neutropénie fébrile observés chez 30 % des patients.
Aucun patient n’a progressé cliniquement en préopératoire.
Le taux de bonne réponse histologique a été de 50 % avec seulement 15 % de très mauvais répondeurs (plus de 50 % de cellules tumorales résiduelles).
Avec un recul médian de 44 mois, aucun des patients bons répondeurs n’a rechuté contre cinq dans le groupe de patients mauvais répondeurs.
Le taux de survie sans récidive à 3 ans est de 74 % et le taux de survie de 90 %.
Ces résultats se comparent favorablement aux deux protocoles précédents proposés chez l’adulte jeune dans cette même situation tumorale à l’IGR avant 1992, le T10 modifié et l’association doxorubicine/cisplatine (AP), aussi bien en termes de réponse histologique qu’en termes de survie sans récidive et de survie globale à 3 ans.
L’analyse statistique de ce groupe homogène de 64 patients montre que les bons résultats obtenus avec le protocole API-AI ne sont pas exclusivement dus à l’augmentation du nombre de patients bons répondeurs mais également à la diminution du nombre de patients très mauvais répondeurs et à l’amélioration de la survie sans récidive des patients mauvais répondeurs, grâce notamment à l’incorporation de l’ifosfamide à fortes doses en postopératoire. Une étude comparative de ces patients adultes pris en charge à l’IGR dans le département de médecine et des jeunes enfants et adolescents pris en charge dans le département de pédiatrie de cette même institution montre que les résultats de l’API-AI (réponse histologique, survie sans récidive et survie globale) sont strictement comparables à ceux observés dans les différentes études prospectives successives menées par la SFOP et contenant toutes du MTX à fortes doses (résultats non encore publiés).
Ces résultats couplés aux données récentes de la littérature suggèrent que l’introduction de l’ifosfamide dans la chimiothérapie des ostéosarcomes induit un gain en termes de survie globale et de survie sans récidive, et que l’absence du MTX n’est pas délétère pour les patients atteints de cette affection.
Un des objectifs de la future étude coordonnée par la Fédération nationale des centres de lutte contre le cancer est de valider ce concept.
2- Formes métastatiques pulmonaires :
Lorsqu’elles surviennent d’emblée, leur traitement est identique à celui des formes localisées en matière de chimiothérapie et de chirurgie de la tumeur.
Après celle-ci, on propose l’exérèse chirurgicale des lésions pulmonaires dans la mesure où elle est techniquement réalisable, seule façon d’obtenir des rémissions à long terme dans ces formes au pronostic sombre. Pour les métastases qui surviennent durant le suivi, le pronostic est directement en rapport avec la date d’apparition et le nombre des lésions existantes.
Selon ces paramètres, le traitement est orienté de préférence vers la chirurgie plus que la chimiothérapie.
C – TRAITEMENT CHIRURGICAL :
L’orientation actuelle du traitement chirurgical des formes localisées est résolument conservatrice ; cependant, il faut savoir rester réaliste et hiérarchiser les priorités du traitement : d’abord sauver la vie, ensuite sauver le membre et ce faisant lui conserver une fonction acceptable.
Cette chirurgie complexe ne se conçoit qu’intégrée dans une stratégie pluridisciplinaire.
En effet, seul un suivi attentif depuis la « simple biopsie » jusqu’à l’acte chirurgical, le plus souvent au décours de la chimiothérapie néoadjuvante, permet de décider quelle est la meilleure tactique chirurgicale.
C’est dire si la collaboration entre les différents membres de l’équipe oncologique est fondamentale, le chirurgien ne devant pas être considéré comme un « sous-traitant » intervenant de façon ponctuelle.
1- Bilan local :
Il doit, si possible, être réalisé avant la biopsie pour éviter que l’imagerie ne soit perturbée par l’oedème postopératoire ou un malencontreux hématome.
Deux examens sont indispensables au chirurgien : les radiographies standards avec mensurations et l’IRM.
Les radiographies permettent d’apprécier la taille, la forme et le volume du segment osseux ou ostéoarticulaire concerné et sont utilisées pour planifier la reconstruction orthopédique lors d’un traitement conservateur.
Cet examen permet seul d’évaluer la solidité osseuse pour prévenir une fracture pathologique par une contention adaptée.
Il permet aussi d’apprécier la consolidation sous traitement médical et orthopédique après cette éventuelle fracture.
L’IRM est l’examen essentiel du bilan d’extension ; elle permet de savoir si le traitement conservateur est possible et quelles sont les limites de la résection.
La discussion avec le radiologue est fondamentale.
L’IRM est très performante sur l’extension osseuse et les parties molles mais reste erratique sur l’envahissement intra-articulaire.
Dans ce cas, les résultats souvent contradictoires peuvent inciter à pécher par excès ou par défaut avec les conséquences fonctionnelles et oncologiques que cela entraîne.
Il faut absolument se souvenir que très souvent le diagnostic est clinique, d’où l’intérêt d’avoir vu le malade vierge de tout traitement, puis au décours de la chimiothérapie néoadjuvante.
2- Méthodes :
* Suppression du membre :
Les amputations ou désarticulations pratiquées pour sarcome ostéogène n’ont rien de particulier.
Leur niveau est déterminé avec autant de rigueur que pour un traitement conservateur, en se méfiant toujours de l’envahissement des parties molles.
La section osseuse s’accompagne d’un prélèvement de moelle au niveau du fût diaphysaire restant, pour s’assurer histologiquement que la section est faite en territoire sain.
Un cas particulier doit être signalé à l’extrémité supérieure du fémur : c’est le risque d’envahissement de l’articulation coxofémorale, celle-ci devant alors être enlevée en bloc.
En fonction de cet envahissement, on peut être obligé de pratiquer une désarticulation inter-ilio-abdominale atypique avec section osseuse transiliaque et transobturatrice, la conservation de l’aile iliaque rendant les appareillages plus faciles que pour une inter-ilioabdominale vraie.
* Conservation partielle du membre avec retournement :
Pour certaines tumeurs du fémur inférieur avec résection en bloc du genou, la jambe restante est retournée de 180° et fixée au fémur diaphysaire restant, plaçant donc la cheville en situation de genou.
L’appareillage est ensuite celui d’une amputation haute de jambe, avec un genou relativement mobile, ce qui donne de meilleurs résultats fonctionnels que l’appareillage d’une amputation à micuisse.
Cependant, il faut noter que cette intervention, bien acceptée en pays anglo-saxon, l’est nettement moins en pays latin à cause de l’esthétique « non naturelle » qu’acquiert le membre inférieur. C’est pourtant une très bonne solution chez le jeune enfant.
Les calculs d’inégalité de croissance permettent d’ajuster la longueur pour que les « deux genoux » soient au même niveau en fin de croissance.
Pour certaines tumeurs de l’extrémité supérieure du fémur, une technique analogue peut être utilisée en fixant le fémur inférieur restant au bassin avec une rotation de 180°, le genou redonnant la flexion et l’extension de la hanche et la cheville faisant office de genou.
* Traitement conservateur :
C’est l’orientation actuelle du traitement local de l’ostéosarcome.
Les résections dans de bonnes conditions de sécurité n’ont été rendues possibles que grâce au traitement médical pré- et postopératoire. L’exérèse monobloc, pour être parfaitement carcinologique, doit enlever l’os et les tissus mous pathologiques, en étant aussi radicale qu’une amputation.
Le bilan d’extension locorégional, souvent complété par une artériographie, a permis de cerner les limites de la tumeur et de décider quel type de résection entreprendre.
En pratique, soit la lésion a des limites d’extension évidentes et on peut, si cela est utile pour conserver une épiphyse ou un cartilage de croissance, tenter une résection limitée mais bien sûr suffisante sur le plan carcinologique, soit les limites restent plus floues, malgré l’apport de l’imagerie (IRM et artériographie) et dans ce cas une résection large s’impose.
L’ostéosarcome est une tumeur relativement « franche », à l’inverse d’autres sarcomes osseux type Ewing ou même chondrosarcome. Avec l’expérience, il est maintenant exceptionnel de passer en zone tumorale.
Encore faut-il se méfier de certains pièges :
– l’extension dans les parties molles doit bien sûr avoir été recherchée, le contrôle des parties molles étant réalisé au mieux en peropératoire par le palper qui apprécie l’envahissement, le doigt étant irremplaçable ; il ne faut pas, par exemple, tenter de conserver une épiphyse sur laquelle s’insèrent des ligaments ou une capsule pathologiques ;
– l’envahissement articulaire : celui-ci est relativement rare, la capsule et le cartilage articulaire, contrairement au cartilage de croissance, semblant une bonne protection contre l’extension tumorale ; en cas de contamination articulaire ou même en cas de doute, l’articulation doit être enlevée avec ses deux versants en monobloc, capsule fermée ; cette tactique, relativement simple à l’épaule, s’avère très sophistiquée au genou et à la hanche ; malheureusement, aucun examen n’est actuellement parfaitement fiable pour détecter cet état de fait ;
– les skip-metastases : il faut s’en méfier dans les tumeurs de gros volume initial à formes ostéocondensantes (2 à 3 % des cas) ; elles peuvent soit sauter l’articulation (atteinte du fémur dans une lésion du tibia supérieur), soit toucher l’os pathologique, mais à distance de la tumeur primitive (lésion du col fémoral sur une tumeur du fémur inférieur) ; leur dépistage repose sur la scintigraphie osseuse et surtout sur l’IRM.
* Reconstruction :
Elle est variable selon l’importance de la résection et son siège diaphysaire ou articulaire.
Le principe essentiel est de faire une reconstruction simple, permettant une récupération fonctionnelle rapide et surtout évitant de retarder la reprise de la chimiothérapie.
Dans certains cas limites, il faut savoir faire une reconstruction « provisoire » et remettre à après la fin du traitement une reconstruction plus sophistiquée mais aussi plus risquée.
Schématiquement, l’attitude actuelle est relativement uniforme :
– en cas de résection diaphysaire, le comblement de la perte de substance osseuse se fait soit par greffe autologue vascularisée ou non, soit par allogreffe ; l’orientation récente étant l’association des deux, permettant un comblement biologique (allogreffe) renforcé par une structure vivante (autogreffe), la séquence opératoire pouvant, en fonction de l’état local, être réalisée en un ou deux temps ;
– en cas de résection articulaire, la préférence va aux prothèses massives ; l’amélioration des modèles actuels les rend plus fiables et pratiques ; à l’épaule et à la hanche, on utilise volontiers des prothèses composites (manchonnées par une allogreffe) qui améliorent la fonction et la stabilité articulaires ; en revanche, au genou, l’adjonction d’allogreffe après résection de l’extrémité inférieure du fémur, péjore le résultat fonctionnel et, après résection de l’extrémité supérieure du tibia, augmente le risque septique.
À part, se pose le problème du cas où, malgré toutes les précautions, on pense avoir ouvert la tumeur.
C’est bien sûr le plus mauvais moment pour décider de remettre en cause le traitement conservateur, bien que le patient ait toujours été prévenu, avant l’intervention, du risque d’amputation.
Il faut, dans ce cas, terminer l’intervention et attendre le résultat histologique de la pièce avant de prendre une nouvelle orientation thérapeutique concernant le traitement local.
Cela dit, si on avait prévu une reconstruction limitée, on peut la réaliser.
En cas de reconstruction sophistiquée ou extensive (longue queue intramédullaire, clou verrouillé, transplant vascularisé…) risquant d’aggraver un éventuel ensemencement, on peut discuter de se limiter à une reconstruction provisoire, quitte à remettre la reconstruction définitive à la fin du traitement adjuvant et avant une éventuelle radiothérapie si celle-ci était décidée.
3- Indications :
En pratique, s’il s’agit d’une localisation à un membre, les indications et contre-indications sont posées en fonction du traitement conservateur proposé en priorité.
Certaines contre-indications à une conservation du membre peuvent apparaître comme d’emblée absolues :
– les tumeurs énormes envahissant les paquets vasculonerveux, dont la résection carcinologique entraînerait un membre paralytique, source de troubles trophiques et sensitifs ;
– les infections persistantes de la biopsie où une résection ferait courir le risque de suites compliquées, retardant la chimiothérapie et amenant finalement à une amputation secondaire ;
– les problèmes cutanés au-dessus des ressources plastiques possibles, interdisant une couverture correcte de la reconstruction ;
– enfin, certains malades vus après une intervention extensive, suite à une erreur diagnostique ou une biopsie irréfléchie ; dans ces cas où il existe un ensemencement des loges musculaires, voire des paquets vasculonerveux, une chirurgie conservatrice n’est plus réalisable en toute sécurité car les limites tumorales deviennent impossibles à cerner.
D’autres contre-indications sont relatives :
– les tumeurs irradiées à des doses supérieures à 30 Gy ont pu parfois être réséquées, mais les complications postopératoires sont plus fréquentes et les résultats fonctionnels toujours médiocres ;
– les tumeurs compliquées de fracture ne sont pas une contreindication absolue ; il faut que le déplacement reste modéré et que l’exérèse puisse emporter non seulement tout le foyer de fracture, mais aussi l’hématome périphérique plus ou moins organisé, obligeant à un sacrifice de parties molles important ;
– l’enfance pose des problèmes difficiles en matière de chirurgie conservatrice ; il ne faut pas raisonner en fonction de l’âge mais en fonction de la perte de croissance prévisible ; schématiquement, audessous de 3 cm, aucun artifice de reconstruction n’est utile, l’égalisation des membres inférieurs est réalisée par épiphysiodèse controlatérale ; entre 3 et 6 cm, on peut, comme c’est le cas au niveau du genou, préserver la croissance du segment ostéoarticulaire conservé en face de la résection, en utilisant un implant spécial provisoire ; celui-ci est uniquement scellé dans l’épiphyse, préservant la croissance du cartilage de conjugaison que la queue prothétique traverse ; on est ainsi ramené au cas précédent ; à l’extrémité supérieure du fémur et de l’humérus, le problème est plus simple en utilisant des prothèses céphaliques pures ; au-delà de 6 à 8 cm, la reconstruction impose un artifice visant à diminuer la perte de croissance tout en essayant de mettre les deux genoux à même niveau en fin de croissance ; dans ce contexte, trois interventions entrent en concurrence : l’amputation, la rotationplastie selon Borgraeve, la prothèse de croissance :
– l’amputation règle tous les problèmes ; malgré son caractère mutilant, elle apporte néanmoins une certaine sérénité au patient et à sa famille, contents d’avoir résolu les difficultés présentes et à venir ;
– le retournement de pied permet de régler l’importance du raccourcissement de façon à ce que les « deux genoux » soient au même niveau en fin de croissance ; il a contre lui son esthétique « chimérique » malgré la pérennité et la qualité de ses résultats fonctionnels ;
– la prothèse de croissance (quels que soient les modèles) n’a encore fait preuve de sa fiabilité, raison pour laquelle les déboires mécaniques restent importants ; il faut bien insister auprès des familles concernées sur l’avenir incertain de cette technique et du résultat final, tant sur la fonction que sur la conservation du membre.
Le choix respectif entre les trois méthodes et dans le cas particulier est une affaire d’expérience et de contexte.
Il faut sûrement passer un long moment avec le patient et ses parents pour définir le choix tactique, aidé par des documents iconographiques (photographies ou vidéo) et des témoignages de patients ayant bénéficié du traitement proposé.
4- Localisations particulières :
* Crâne et face :
Elles sont très rares ; mais souvent la résection locale ne peut être effectuée réellement en bloc et une irradiation complémentaire peut être nécessaire, voire utilisée systématiquement.
* Bassin et sacrum :
Malgré la possibilité de faire, à ce niveau, des exérèses carcinologiquement satisfaisantes, le pronostic de ces localisations dans le cadre de l’ostéosarcome de haut grade reste sombre, du fait de l’important volume tumoral et de l’envahissement des plexus veineux sacrés et hypogastriques.
Seule une réponse optimale à la chimiothérapie peut améliorer le contrôle local et général de cette localisation.
* Omoplate :
Il s’agit d’une localisation rare mais avec souvent un volume tumoral important ; l’extension à la paroi thoracique est tout à fait exceptionnelle en dehors des formes radio-induites.
La difficulté pour cet os d’avoir une imagerie correcte de l’envahissement endomédullaire impose le plus souvent de faire une scapulectomie totale avec une reconstruction suspendant l’humérus restant à la clavicule et un résultat fonctionnel médiocre en découle, avec cependant un membre supérieur fonctionnel au coude et à la main.
* Localisations vertébrales :
Les ostéosarcomes de forme commune y sont relativement rares et il s’agit le plus souvent de formes différenciées ou radio-induites et donc faiblement sensibles à la chimiothérapie adjuvante.
Ce facteur, associé à la localisation où il est très difficile de faire une chirurgie large et parfaitement carcinologique, explique le mauvais pronostic et les difficultés à obtenir un contrôle local correct.
Cependant, la vertébrectomie totale après confirmation histologique reste le meilleur moyen de donner une chance de guérison à ce type de patient.
* Localisations à la paroi thoracique :
Les localisations costales sont moins fréquentes que dans le cadre du sarcome d’Ewing.
Cependant, elles posent un problème majeur de prise en charge propre à la chirurgie thoracique.
En effet, dans les sarcomes costaux de haut grade, il est impératif d’opérer ces patients après une histologie préalable et en évitant de faire des pariétectomies localisées, l’intervention chirurgicale devant absolument enlever la totalité de la côte malade ainsi que les espaces intercostaux sus- et sous-jacents en incluant les côtes adjacentes.
Pour l’articulation costovertébrale, dans les formes à localisation postérieure, il est impératif de faire une résection extra-articulaire emmenant la transverse et une partie du corps vertébral.
En cas d’envahissement de la gouttière pariétovertébrale, ce type de localisation oblige parfois, pour obtenir des marges d’exérèse suffisantes, à réaliser d’emblée une hémivertébrectomie correspondant aux articulations costovertébrales concernées, associée à la pariétectomie thoracique.
D – RADIOTHÉRAPIE :
Contrairement à la chirurgie et à la chimiothérapie, la radiothérapie (RTE) a peu de place dans la prise en charge thérapeutique des ostéosarcomes du fait de l’histoire naturelle de cette tumeur et de sa relative radiorésistance.
Les rechutes locales après chimiothérapie néoadjuvante et chirurgie sont en effet rares dans les ostéosarcomes des membres (moins de 10 %) et ne justifient donc pas d’un traitement local complémentaire.
Avant l’introduction de la chimiothérapie dans les années 1970, la RTE faisait partie du traitement local afin d’éviter l’amputation chez des patients qui de toute façon mouraient le plus souvent d’une diffusion métastatique pulmonaire à court terme.
À cause des particularités radiobiologiques de l’ostéosarcome, les doses d’irradiation nécessaires étaient élevées et pouvaient entraîner des complications trophiques à long terme chez les quelques survivants.
Les indications de RTE sont donc actuellement peu nombreuses.
1- Ostéosarcome des os de la face (maxillaire et mandibule) :
La RTE externe et/ou la curiethérapie interstitielle pré- ou postopératoire permettent d’améliorer la survie à long terme de ces patients qui meurent pour la plupart d’évolution locale.
2- Ostéosarcomes inopérables (bassin, rachis) ou rechutes locales :
La RTE peut être proposée en cas de rechute locale isolée.
En ce qui concerne les ostéosarcomes inopérables, il s’agit le plus souvent de traitements palliatifs, avec un bénéfice observé chez 40 à 50 % des patients.
En raison de la radiorésistance de ces lésions, d’autres techniques de radiothérapie ont pu être évaluées dans des groupes souvent limités de patients, certaines utilisant des neutrons, des ions lourds ou encore la radiothérapie peropératoire ou la radiothérapie conventionnelle avec des radiosensibilisants (bromodéoxyuridine, l’iododéoxyuridine ou chimiothérapie concomitante) avec des résultats intéressants mais pouvant entraîner des séquelles trophiques parfois importantes.
La protonthérapie peut être indiquée pour certaines localisations. Afin de diminuer le risque de rechute métastatique pulmonaire, une irradiation des deux poumons à des doses variant entre 15 et 20 Gy a pu être proposée avec des résultats encourageants.
Un essai randomisé à trois bras a donc comparé une chimiothérapie adjuvante à une radiothérapie pulmonaire prophylactique, ou à l’association des deux.
Il n’y avait pas de différence entre les différents bras. Néanmoins, si la chimiothérapie fait actuellement partie du traitement standard des ostéosarcomes, la RT prophylactique pulmonaire ne peut être préconisée en dehors d’essais.
Conclusion :
Bien que l’ostéosarcome soit la tumeur maligne primitive osseuse la plus fréquente, myélome exclu, la grande diversité aussi bien clinique, radiologique, histologique qui le caractérise fait de chaque patient un cas particulier.
Son pronostic a été radicalement amélioré grâce à l’introduction de la polychimiothérapie.
Sa prise en charge, parfaitement codifiée, comporte une biopsie chirurgicale à visée diagnostique, une chimiothérapie d’induction, une exérèse chirurgicale suivie d’une chimiothérapie adjuvante et parfois une radiothérapie.
Le traitement veille à la conservation de la fonction du membre atteint tout en assurant une exérèse carcinologique satisfaisante.
Plus que la qualité propre de chaque discipline médicale impliquée dans sa prise en charge, c’est leur parfaite symbiose qui régit la qualité du résultat.
L’ostéosarcome constitue ainsi un modèle de tumeur pour la prise en charge multidisciplinaire.







")



{kind=link}