



















Introduction :
Les granulomes palissadiques (GP) sont un cadre anatomopathologique rassemblant des affections de nature variée.
Leur point commun est une image histologique appelée « granulome palissadique ».
Les plus fréquentes sont :
– le granulome annulaire (GA) (et le granulome actinique) ;
– la nécrobiose lipoïdique (et la granulomatose disciforme de Miescher) ;
– les nodules rhumatoïdes (et nodules rhumatismaux et ceux de la maladie de Still) ;
– le xanthogranulome nécrobiotique ;
– la dermatite interstitielle granulomateuse ;
– les granulomes nécrosants extravasculaires.
 Certaines maladies granulomateuses, habituellement non palissadiques, peuvent exceptionnellement former des palissades : réaction à corps étranger, dermatoses infectieuses.
Certaines maladies granulomateuses, habituellement non palissadiques, peuvent exceptionnellement former des palissades : réaction à corps étranger, dermatoses infectieuses.
Le diagnostic peut être aisé cliniquement, confirmé par l’examen histologique.
Dans bon nombre de cas, cependant, seule une confrontation des aspects histologiques observés aux lésions cliniques et au contexte pathologique permet une approche diagnostique satisfaisante.
Définition :
Les dermatoses regroupées sous le terme de « granulome palissadique » sont caractérisées par la présence d’un infiltrat inflammatoire dermohypodermique majoritairement constitué d’histiocytes (« granulome histiocytaire » appelé simplement « granulome » par les Anglo-Saxons).
Ils se regroupent et s’alignent pour former des « palissades » plus ou moins bien définies autour ou au contact de plages d’altération du tissu conjonctif appelée « nécrobiose » dont la nature exacte n’est pas connue.
Ces plages montrent une détérioration des faisceaux collagènes (fragmentation, épaississement avec hyalinisation) et parfois élastiques, mais également une régénération et une apparition de néofaisceaux.
La disposition palissadique des histiocytes doit être distinguée d’une simple disposition interstitielle, sans organisation particulière, fréquemment observée dans ces pathologies.
Si celle-ci n’est pas associée à des altérations conjonctives, elle oriente alors vers d’autres affections.
Quatre types de GP peuvent être individualisés en fonction de l’architecture de la lésion :
– GP classique : les histiocytes s’alignent pour former une « palissade » entourant une plage de nécrobiose (le plus souvent éosinophile) ;
– granulome tuberculoïde avec nécrobiose : les histiocytes forment de véritables granulomes tuberculoïdes (ou épithélioïdes et gigantocellulaires) à proximité d’un tissu conjonctif le plus souvent peu altéré ;
– granulome interstitiel avec rosettes : les histiocytes prennent une disposition interstitielle sans organisation particulière, entre les faisceaux collagènes dermiques, qui sont peu altérés et se regroupent parfois en petites palissades entourant un ou quelques faisceaux collagènes altérés (éosinophiles ou basophiles), appelés « rosettes » ;
– granulome nécrosant extravasculaire : les histiocytes forment des palissades, souvent moins nettes que dans la forme classique, entourant une plage de nécrose basophile, formant parfois des images en « flammèches ».
Ces types histologiques sont caricaturaux et correspondent aux aspects typiques qui, lorsqu’ils existent, permettent un diagnostic aisé.
Souvent l’aspect est moins caricatural et une disposition vaguement palissadique des histiocytes, l’existence de signes d’altérations du tissu conjonctif, doivent être recherchés avec soin au sein de tout infiltrat inflammatoire granulomateux interstitiel.
Granulome annulaire :
Cette maladie inflammatoire cutanée bénigne, décrite depuis plus d’un siècle, reste d’étiologie inconnue.
Son évolution est spontanément régressive.
L’éruption est, de façon caractéristique, papuloérythémateuse, annulaire, peu ou pas prurigineuse, touchant habituellement les mains et les pieds.
A – ÉPIDÉMIOLOGIE :
Plus fréquente chez la femme (sex-ratio 3/1), cette dermatose peut toucher toutes les tranches d’âge, l’enfant comme le sujet âgé.
Elle est cependant préférentiellement observée chez les moins de 30 ans. Une prédisposition familiale a été suggérée devant des cas touchant des jumeaux ou plusieurs générations d’une même famille.
Dans 75 % des cas, le GA disparaît spontanément en 2 mois à 2 ans après son apparition.
Cependant, 40 % des enfants peuvent présenter des récidives, souvent de même localisation. Des pics saisonniers ont été observés au printemps et à l’automne.
Il n’existe pas d’altération biologique.
B – ÉTIOLOGIE :
Son étiologie demeure obscure. Certains GA surviennent après diverses infections (virales : virus de l’immunodéficience humaine [VIH], virus de l’hépatite C [VHC], virus Epstein-Barr, herpes simplex et varicelle ; fungiques ; mycobactériennes ; borrélienne) après des réactions d’hypersensibilité ou des traumatismes.
Le rôle d’un traumatisme initial avec nécrobiose dermique secondaire est évoqué dans les GA survenant après piqûres d’insectes, test à la tuberculine, irritation vestimentaire.
Cette nécrobiose pourrait être également liée à la libération anormale d’enzymes lysosomiales par les histiocytes.
Une réaction d’hypersensibilité de type IV liée aux lymphocytes T-helper rencontrés en périphérie de la lésion a été évoquée.
En effet, lorsqu’ils sont activés, ces lymphocytes T peuvent produire des cytokines intervenant dans la synthèse du collagène (transforming growth factor [TGF]-b, fibroblast stimulating interleukins [IL]1, immune modulating IL2) ou de sa destruction par des collagénases (immune modulating IL2).
Ce type de mécanisme est envisagé pour expliquer la survenue de GA au cours du syndrome de l’immunodéficience acquise (sida), en particulier dans d’anciens sites d’infections herpétiques.
Chez l’enfant, il n’est pas associé à des pathologies systémiques.
Chez l’adulte, une possible relation entre le GA disséminé et le diabète a été suggérée par certains auteurs.
Cette association reste controversée : les dernières études épidémiologiques semblent écarter toute relation avec le diabète de type 2 alors que la fréquence du diabète insulinodépendant pourrait être augmentée, surtout dans les formes localisées de GA.
La survenue de GA après une exposition solaire intense n’a pas pu être reproduite en lumière artificielle.
C – CLINIQUE :
Quatre tableaux cliniques sont habituellement décrits : GA localisé, généralisé, perforant ou sous-cutané.
1- Forme localisée :
C’est la plus fréquente, avec la survenue d’un nombre limité de papules de 2 à 4mm, de couleur peau normale, érythémateuse ou brune, de disposition arciforme ou annulaire, sans signe épidermique.
Les papules solitaires existent mais les lésions sont habituellement multiples.
Elles peuvent confluer pour former de larges plaques annulaires.
Le centre des lésions annulaires peut être déprimé et pigmenté.
Les lésions sont non douloureuses et non prurigineuses.
Elles sont localisées aux extrémités.
Dans 60 % des cas, elles touchent les mains et les bras, les jambes et les pieds dans 20 %, les deux dans 7 % des cas.
Épargnant habituellement les paumes, les plantes et les muqueuses, des atteintes buccales ont été cependant rapportées au cours du sida.
L’évolution spontanément résolutive survient chez environ 50 % des patients ; cependant, elles peuvent persister près de 2 ans.
De nombreuses dermatoses peuvent être confondues avec le GA.
Dans les formes localisées, les principaux diagnostics différentiels sont : le lichen plan annulaire, l’erythema elevatum diutinum, la syphilis et certaines formes unilatérales de nécrobiose lipoïdique qui peuvent être indiscernables d’un GA.
2- Forme généralisée :
Plusieurs centaines de petites papules de 1 à 2mm, isolées ou coalescentes, forment plus rarement de petites plaques annulaires, érythématoviolines ou lilacées à bordure à peine papuleuse, disséminées, de taille variable, intéressant le tronc, le cou et la partie proximale des membres.
Le cuir chevelu, les paumes et les plantes ne sont pas habituellement touchés.
Concernant presque exclusivement les adultes, sa régression spontanée est moins fréquente, rare avant 3 à 4 ans.
Cette forme semble être liée à un groupe human leukocyte antigen (HLA) Bw35 ou A29.
Elle est également la forme la plus fréquemment rencontrée chez le patient VIH.
Les formes papuleuses multiples et diffuses font discuter un lichen plan, une sarcoïdose ou une syphilis.
Une borréliose (érythème chronique migrateur), un érythème annulaire centrifuge, des morphées débutantes au stade inflammatoire, un mycosis fongoïde doivent être éliminés devant la présence de plaques.
3- Granulome annulaire perforant :
Il est rare, plus fréquent chez l’enfant que chez l’adulte.
Il est caractérisé par de petites papules superficielles ombiliquées, comportant une croûte ou une cicatrice centrale, touchant surtout les doigts et les mains (penas).
Les lésions s’aggraveraient pendant l’été et s’amélioreraient en hiver.
Un molluscum contagiosum et certaines dermatoses perforantes telles que l’élastose perforante serpigineuse, font partie du diagnostic différentiel.
4- Forme sous-cutanée :
Elle est surtout rencontrée chez l’enfant.
Elle comporte des lésions plus grandes, nodulaires, d’apparition rapide.
Elles sont non douloureuses, fermes et non mobiles, mesurant de 1 à 5 cm.
Touchant les extrémités, particulièrement les aires prétibiales, elles atteignent également les mains, le cuir chevelu, les régions périorbitaires et les fesses.
Certaines lésions apparaissent fixées au tissu périosté.
Elles sont, de façon caractéristique, isolées ou parfois regroupées en petits amas. Un quart des patients ont aussi un aspect de GA localisé.
La résolution spontanée est la règle comme dans la forme localisée, mais les lésions peuvent récidiver après plusieurs années.
Le diagnostic est cliniquement difficile, souvent confondu avec un processus tumoral.
D – HISTOLOGIE :
Le GA est caractérisé par un infiltrat inflammatoire du derme superficiel et moyen, histiocytaire associé à quelques lymphocytes péricapillaires.
Ces histiocytes se regroupent plutôt en foyer.
Ils prennent, soit une disposition interstitielle sans organisation visible, soit forment typiquement des palissades autour d’un foyer de dégénérescence éosinophile, généralement incomplète, du collagène dermique associé à des dépôts de substance mucineuse.
Tous les intermédiaires entre ces deux extrêmes sont possibles sur la même biopsie ou sur des lésions différentes.
Occasionnellement, les histiocytes peuvent former des granulomes tuberculoïdes.
Les dépôts mucineux sont granuleux, basophiles, plus ou moins bien visibles sur les colorations standards.
Ils doivent être recherchés par une coloration spéciale (exemple : bleu alcian).
Rarement, quelques polynucléaires neutrophiles ou éosinophiles sont observés, parfois en leucocytoclasie.
Les cellules géantes, lorsqu’elles sont présentes, sont peu nombreuses.
Dans la forme perforante, la lésion est très superficielle, s’accompagnant d’une ulcération épidermique.
Dans la forme sous-cutanée, le granulome est typiquement palissadique, entourant une vaste plage de dégénérescence collagénique.
Des mitoses et atypies cytologiques des histiocytes ont été rapportées par certains auteurs.
Il est à noter qu’il semble exister une certaine corrélation entre les différentes formes cliniques de GA et les différents types histologiques : les GA localisés, perforants et sous-cutanés sont le plus souvent typiquement palissadiques (seule leur situation plus ou moins profonde dans le derme diffère).
Les GA généralisés en petites papules sont volontiers interstitiels avec des rosettes alors que ceux en petites plaques sont tuberculoïdes.
Le diagnostic histologique des formes classiques ne pose pas de difficulté.
Dans les formes interstitielles, une borréliose, une morphée ou même une variété récemment individualisée de mycosis fongoïde dite « interstitielle » peuvent se discuter.
La présence de plasmocytes est évocatrice d’une borréliose.
Un infiltrat plus lymphocytaire qu’histiocytaire associé à une sclérose collagénique plus ou moins marquée sont des arguments en faveur d’une morphée.
L’épidermotropisme lymphocytaire, même s’il se limite parfois à un simple alignement des lymphocytes le long de la basale épidermique, représente l’argument diagnostique clé en faveur du mycosis fongoïde.
Les formes tuberculoïdes font discuter une sarcoïdose (la disposition interstitielle du granulome, la présence de mucine permettent d’écarter ce diagnostic) ou plus rarement une forme dite « granulomateuse » de mycosis fongoïde, où les lymphocytes tumoraux sont mêlés à un infiltrat histiocytaire interstitiel et tuberculoïde.
E – FORME PARTICULIÈRE :
Certains auteurs distinguent les GA des granulomes actiniques alors que d’autres considèrent que les granulomes actiniques sont simplement des GA qui surviennent sur peau ensoleillée.
Le diagnostic de ces granulomes actiniques est anatomoclinique.
Touchant l’homme et la femme de façon indifférente après 30 ou 40 ans, ils surviennent surtout sur le nez, la face, le thorax et les bras.
Un phototype clair et l’âge avancé seraient des facteurs favorisants.
Ils débutent par l’apparition d’une ou plusieurs petites papules érythémateuses qui évoluent pour former une plaque de plusieurs centimètres, parfois annulaire, dont le centre peut être atrophique et hypopigmenté.
En histologie, la biopsie des berges permet de distinguer trois zones :
– en dehors de la plaque : des signes d’héliodermie : une dégénérescence élascéinique du derme papillaire et une atrophie épidermique ;
– sur les berges : une réaction granulomateuse riche en cellules géantes avec élastophagie, sans nécrobiose, sans palissade histiocytaire, associée à des plasmocytes et des dépôts mucineux ou lipidiques ;
– au centre de la lésion : une absence de tissu élastique normal ou dégénéré.
Survenant dans des zones où il existe une dégénérescence élascéinique (derme superficiel), ils se caractérisent par une élastophagocytose avec, pour conséquence une disparition des fibres élastiques.
Certains critères permettraient une distinction entre les GA et les granulomes actiniques.
Les granulomes actiniques surviennent volontiers dans le derme superficiel, au niveau de l’élastose solaire.
Ils sont plus volontiers tuberculoïdes ou interstitiels que palissadiques.
Ils ne s’accompagnent pas de dépôt de mucine et seraient plus riches en cellules géantes que le GA.
F – TRAITEMENT :
Aucun traitement n’est habituellement nécessaire.
Le retentissement esthétique, la dissémination des lésions ont conduit à l’utilisation de diverses modalités thérapeutiques, aucune n’ayant prouvé son efficacité.
Les traitements locaux : cryothérapie, laser, excision chirurgicale, injections intralésionnelles de corticoïdes ; les traitements généraux : anti-inflammatoires non stéroïdiens (AINS), thalidomide, vitamine E, antipaludéens…peuvent être utilisés en fonction de chaque cas particulier.
Nécrobiose lipoïdique :
A – DÉFINITION :
Décrite en 1929 par Oppenheim sous le nom de dermatitis atrophicans diabetica, c’est Urbach, en 1932, qui rapporta ces lésions sous leur appellation définitive de necrobiosis lipoidica diabetorum.
Cette dermatose d’étiologie inconnue est classiquement associée au diabète bien qu’elle reste rare dans la population diabétique puisque seuls 0,7 % des patients diabétiques sont également porteurs d’une nécrobiose lipoïdique.
En effet, il est communément admis qu’environ les trois quarts des patients porteurs d’une nécrobiose lipoïdique vont développer un diabète.
Cette association, selon une étude récente, ne serait pas aussi forte puisque O’Toole et al signalent, en 1999, que seulement 11 % de leur patients atteints d’une nécrobiose lipoïdique sont diabétiques.
Des cas de nécrobiose lipoïdique ont été également rapportés au cours de maladie de Crohn, de rectocolite ulcérohémorragique, après dérivation jéjunale chirurgicale (bypass jejunal) et au cours de pathologie thyroïdienne auto-immune (thyroïdite d’Hashimoto et maladie de Basedow).
Elle touche l’adulte jeune, surtout les femmes (sex-ratio : 3/1).
B – CLINIQUE :
Les lésions typiques se développent sur les régions prétibiales.
Des localisations plus inhabituelles aux chevilles, mollets, cuisses, creux poplités et pieds sont possibles.
Il s’agit d’une « dermite sclérodermiforme atrophiante ».
Les lésions débutent sous forme de petites papules érythémateuses, fermes, qui progressivement confluent et s’élargissent pour former des plaques.
Les plaques s’étendent lentement devenant ovoïdes, parfois squameuses à bordure violacée, indurée, à centre atrophique jaunâtre et télangiectasique.
Ces plaques sont habituellement multiples et bilatérales mais peuvent être uniques et unilatérales.
D’évolution chronique, elles peuvent adhérer au plan osseux profond ou s’ulcérer après un traumatisme minime.
Ces ulcérations sont particulièrement réfractaires aux traitements médicaux et chirurgicaux.
Elles s’accompagnent d’un risque de surinfection et de survenue de douleurs.
Une rémission survient dans 15 % des cas après 1 à 35 années d’évolution au prix de cicatrices atrophiques résiduelles.
Dans environ 15 % des cas, les lésions sont présentes dans d’autres localisations (dos des mains, doigts, avant-bras, tronc, visage, cuir chevelu), le plus souvent associées aux lésions prétibiales.
Les lésions sont alors plus papuleuses et nodulaires sans atrophie, se rapprochant d’un GA.
Des phénomènes de Koebner ont été rapportés.
La granulomatose disciforme de Miescher est considérée comme une variante clinique particulière par son atteinte isolée du visage ou du cuir chevelu, chez des sujets habituellement non diabétiques.
Il s’agit d’une plaque érythémateuse ou brune à bordure annulaire ou serpigineuse, non infiltrée.
Cette forme a été individualisée du fait de la relative absence de nécrobiose en histologie.
Elle pourrait être également rapprochée du granulome de O’Brien.
Le GA représente le principal diagnostic différentiel. Une sarcoïdose, un xanthome, une morphée, un pyoderma gangrenosum, une syphilis tertiaire, une radiodermite, une mycobactériose atypique peuvent être également discutés.
C – HISTOLOGIE :
L’épiderme est normal, atrophique ou hyperkératosique.
Toute l’épaisseur du derme ou ses deux tiers inférieurs sont occupés par un granulome palissadique typique ou dégradé, une nécrobiose et une sclérose à des degrés variables.
S’y associent quelques lymphocytes péricapillaires et parfois des plasmocytes en profondeur.
L’aspect histologique est cependant fonction du stade évolutif des lésions et du site de la biopsie.
Il est souvent difficile à distinguer d’un GA.
En faveur d’une nécrobiose lipoïdique, on retient : le caractère plus profond et diffus des lésions, pouvant atteindre l’hypoderme et respectant le derme superficiel, la tendance à l’étalement et à l’horizontalisation des foyers de nécrobiose, l’absence ou la rareté des dépôts de mucine, la présence d’altérations vasculaires (turgescence endothéliale, fibrose de la paroi avec hyalinisation et présence de dépôts à acide périodique de Schiff [PAS] +), de dépôts lipidiques (extracellulaires ou intracellulaires sous forme d’histiocytes spumeux), la richesse de l’infiltrat en cellules géantes.
D – TRAITEMENT :
L’évolution de la nécrobiose lipoïdique n’est pas liée au contrôle glycémique.
Différents traitements semblent améliorer l’évolution des lésions : la corticothérapie locale intralésionnelle ou occlusive, générale dont l’utilisation est limitée par les effets secondaires, la puvathérapie, ou la greffe dermique peuvent être envisagées.
Certains auteurs ont rapporté récemment des effets bénéfiques de la chloroquine.
Nodules rhumatoïdes :
Ils surviennent chez 20 à 30 % des patients atteints d’une polyarthrite rhumatoïde ; ils sont plus volontiers associés aux formes sévères, aux formes destructrices séropositives et aux vascularites rhumatoïdes.
Ils siègent surtout sur les faces d’extension des membres, en regard des articulations métacarpophalangiennes et interphalangiennes proximales.
L’atteinte histologique est dermique profonde et hypodermique recouvrant l’os, en particulier dans des zones de traumatismes mineurs répétés : olécrane, surface ulnaire de l’avantbras, face d’extension des mains, épaules, tubérosités ischiatiques, sacrum, genoux, pieds, région postérieure du cuir chevelu (mais également arête nasale, pavillon de l’oreille, yeux, coeur, poumons, plèvre, paroi abdominale, péritoine, capsule splénique et larynx).
Il s’agit de petits nodules sous-cutanés en relief, fermes, indolores, sans coloration particulière, fixes ou mobiles, mesurant au maximum plusieurs centimètres de diamètre.
Parfois, la lésion s’ulcère, se surinfecte ou se draine. Rarement, l’os sous-jacent est érodé ou un nerf comprimé.
Ces nodules peuvent précéder de plusieurs années l’apparition des lésions articulaires.
Ces nodules se développent insidieusement, persistant de façon chronique, pouvant néanmoins régresser, surtout si le mécanisme irritatif disparaît.
Histologiquement, les nodules sont hypodermiques ou dermiques profonds, se caractérisant par trois zones distinctes : une zone centrale de nécrose fibrinoïde collagénique entourée par une palissade histiocytaire (avec quelques cellules géantes de type Muller) et enfin plus en périphérie, un tissu inflammatoire de granulation à type de bourgeon charnu, des infiltrats lymphoïdes péricapillaires et une fibrose.
Le diagnostic différentiel se pose surtout avec les formes profondes de GA (pseudonodules rhumatoïdes).
L’absence de dépôts mucineux, la présence d’une fibrose d’enkystement et de nombreuses cellules géantes peuvent être des arguments histologiques orientant vers un nodule rhumatoïde.
C’est surtout l’existence d’une polyarthrite rhumatoïde qui permet de retenir le diagnostic.
De multiples mécanismes pathogéniques, tels que des traumatismes, la libération d’enzymes protéolytiques, une vascularite liée aux complexes immuns, une réponse immunitaire spécifique liée aux cellules T ou une prédisposition génétique ont été suggérés comme possibles facteurs responsables de l’apparition des nodules rhumatoïdes.
Rarement, ces nodules ont été associés à certaines maladies systémiques : le lupus érythémateux systémique (5 %-7 % des patients porteurs d’un lupus), spondylarthrite ankylosante séronégative et syndrome de Jaccoud.
Nodules rhumatismaux et ceux de la maladie de Still :
Les rhumatismes articulaires aigus touchent surtout les enfants des pays en voie de développement et sont exceptionnels dans nos régions.
Liés à une infection à streptocoque bêtahémolytique du groupe A, les nodules rhumatismaux (ou nodules de Meynet) sont plutôt observés dans les formes sévères avec atteinte cardiaque.
Ils se présentent comme des nodules sous-cutanés, fermes, indolores, siégeant plus particulièrement à proximité des articulations, au contact des proéminences osseuses (coude, genou).
Ils sont de plus petite taille et ont une évolution plus courte que les nodules rhumatoïdes.
Histologiquement, ils se rapprochent également des nodules rhumatoïdes.
Ils s’en distingueraient par une délimitation moins nette, l’intensité des dépôts fibrinoïdes, l’existence de remaniements oedémateux, la discrétion de la réaction inflammatoire, une disposition palissadique moins évidente, l’absence ou la discrétion de la fibrose.
Des nodules semblables ont été rapportés dans la maladie de Still.
Xanthogranulome nécrobiotique :
Cette dermatose rare, décrite en 1980, se présente sous la forme de nodules et plaques infiltrés, de coloration jaune à brune, parfois ulcérés ou cicatriciels, atteignant de façon très évocatrice les régions périorbitaires et plus rarement le tronc, la face et les membres.
Des cas exceptionnels d’atteinte cutanée généralisée ou d’atteinte systémique (poumon, myocarde, ovaire, rein, foie et rate) ont été rapportés.
Elle s’accompagne dans plus de 80 % des cas d’une paraprotéine de type immunoglobuline (Ig)G kappa de signification indéterminée ou dans le cadre d’un myélome, d’un syndrome lymphoprolifératif ou d’une cryoglobulinémie.
Une leucopénie, une hypocomplémentémie, une augmentation de la vitesse de sédimentation peuvent être retrouvées.
L’évolution est lentement progressive avec augmentation de la taille et du nombre des lésions ; leur régression spontanée est possible.
Histologiquement, le derme et l’hypoderme sont le siège d’une réaction inflammatoire granulomateuse plurifocale séparée par des zones de nécrobiose du tissu conjonctif.
Généralement interstitiel, le granulome peut parfois adopter ici une disposition palissadique au contact de ces plages de nécrose.
Le granulome est riche en macrophages spumeux, en cellules géantes de Touton (aux noyaux disposés en cercle dans un cytoplasme spumeux) ou de type Muller (à corps étranger : aux noyaux dispersés dans un cytoplasme éosinophile) de forme parfois anguleuse « bizarre ».
Des empreintes de cristaux de cholestérol sont fréquemment observées au contact des plages de nécrose.
Des plasmocytes, lymphocytes et follicules lymphoïdes sont moins fréquents.
Les modalités thérapeutiques ne sont pas définies : divers traitements ont été tentés, cytotoxiques (chlorambucil, melphalan), plasmaphérèse ou corticothérapie orale à forte dose.
Dermatite interstitielle granulomateuse :
La dermatite interstitielle granulomateuse (DIG) est un nouveau cadre anatomoclinique récemment individualisé, caractérisé par la présence d’un granulome interstitiel pouvant focalement adopter une disposition palissadique qui justifie ici sa description.
Cette entité semble regrouper :
– la dermatite interstitielle granulomateuse avec arthrite ;
– la dermatite interstitielle granulomateuse liée aux médicaments.
En 1993, Ackerman et al ont décrit une dermatose rare sous le terme de « dermatite interstitielle granulomateuse avec cordons cutanés et arthrite ».
Des lésions identiques avaient déjà été rapportées sous différentes appellations : « bande linéaire sous-cutanée dans la polyarthrite rhumatoïde », « GA linéaire », « nodules rhumatoïdes linéaires », « dermatose en rails ».
Touchant préférentiellement les femmes, elle se traduit par l’apparition progressive et asymptomatique de cordons verticaux, bilatéraux et parfois symétriques (signe de la corde), des plis axillaires mais aussi des parois thoraciques latérales et de l’abdomen et serait pathognomonique.
D’autres lésions moins évocatrices ont été décrites sous le terme de « dermatite interstitielle granulomateuse avec plaques » : multiples nodules ou plaques violacées à brunes (mesurant jusqu’à 18 cm) annulaires, à bords nets, plus ou moins infiltrés des parois thoraciques et des cuisses, bilatéraux et parfois symétriques.
Cette éruption est associée à des polyarthralgies (atteinte symétrique des doigts, des poignets, des coudes ou des épaules) et survient dans un contexte auto-immun, le plus souvent mal étiqueté.
Facteurs rhumatoïdes et anticorps antinucléaires peuvent être retrouvés.
Certains cas sont associés à une véritable polyarthrite rhumatoïde, un lupus érythémateux systémique, un diabète ou une thyroïdite.
Ces arthrites accompagnent, précèdent ou suivent les lésions cutanées.
La fréquence des différentes formes d’expression clinique et de leur association à des maladies auto-immunes n’est pas connue.
L’évolution est marquée par une alternance de poussées et de rémissions.
Ces lésions disparaissent spontanément en 6 à 12 mois. L’histologie est caractérisée par la présence d’un infiltrat histiocytaire interstitiel diffus et dense du derme réticulaire, adoptant une disposition en « bande ».
Il s’y associe des lymphocytes péricapillaires, des polynucléaires neutrophiles ou éosinophiles.
Ces derniers sont surtout abondants à proximité de GP particuliers appelés « rosettes » : les histiocytes forment de petites palissades autour d’un ou de quelques faisceaux collagènes altérés basophiles.
Ailleurs, les altérations du collagène sont discrètes.
Des granulomes de type nécrosant extravasculaire ont été également rapportés.
Les cellules géantes sont généralement peu nombreuses. Il n’existe pas de mucine, pas de vascularite.
Les histiocytes ont un noyau souvent volumineux et pléomorphe.
Des mitoses sont parfois observées ainsi que des lymphocytes atypiques.
La dermatite interstitielle granulomateuse liée aux médicaments, décrite par Magro et al en 1998, semble être une forme particulière.
Elle se distingue par son étiologie médicamenteuse prouvée par la disparition des signes cutanés après arrêt du médicament causal et par quelques particularités cliniques et histologiques.
Les classes médicamenteuses incriminées ne diffèrent pas des classes habituellement en cause dans les toxidermies (inhibiteurs calciques, bêtabloquants, hypolipémiants, inhibiteurs de l’enzyme de conversion de l’angiotensine…).
Le délai d’apparition de l’éruption est très variable (de 4 semaines à plusieurs années).
Il s’agit de plaques asymptomatiques érythémateuses ou violacées des plis, des faces internes des avant-bras ou des régions intermammaires, évoquant cliniquement un GA ou un mycosis fongoïde.
En histologie, le granulome interstitiel comporte des images en rosettes et également des granulomes nécrosants extravasculaires.
Il présente la particularité d’être associé, dans certains cas, à :
– une altération basale épidermique : dermatose vacuolaire d’interface, voire dermatose lichénoïde (avec présence d’altération kératinocytaire basale à type de vacuolisation ou de dyskératose et exocytose lymphocytaire) ;
– des images d’élastophagie, avec de nombreuses cellules géantes.
Le diagnostic différentiel se pose sur le plan clinique et histologique avec une forme interstitielle de mycosis fongoïde.
Leur distinction est d’autant plus difficile que des lymphocytes sézariformes sont présents dans environ 50 % des cas de dermatite interstitielle granulomateuse.
La disparition des lésions après éviction du médicament responsable semble être le critère le plus fiable pour distinguer ces deux entités.
Granulomes nécrosants extravasculaires :
Ces granulomes, initialement décrits dans la granulomatose allergique sous le nom de « granulome de Churg et Strauss », ont été également appelés : granulome allergique, granulome de Winkelmann, papule rhumatoïde, nécrobiose superficielle ulcérante, dermatite neutrophilique rhumatoïde, phénomène de Churg et Strauss.
« Granulome nécrosant extravasculaire » est un synonyme proposé par Finan en 1983 pour éviter toute confusion avec la granulomatose de Churg et Strauss. Des lésions identiques sont en effet observées dans d’autres maladies systémiques (lupus érythémateux, maladie de Wegener, périartérite noueuse, polyarthrite rhumatoïde, endocardite bactérienne, hépatite chronique active, vascularite, syndromes lymphoprolifératifs, colite inflammatoire et sida).
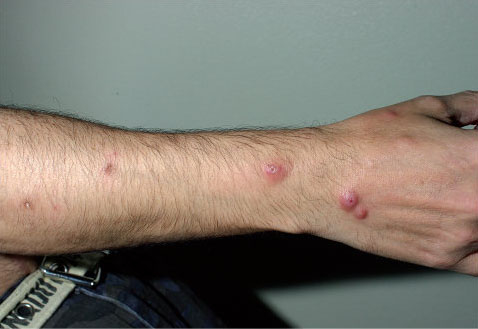
Il s’agit de façon caractéristique de papules ou de petits nodules ombiliqués, de couleur peau normale ou rouge violacé, prédominant sur les coudes et les doigts.
Les lésions sont en règle symétriques et multiples.
Le mécanisme d’apparition de ces lésions n’est pas connu.
Leurs localisations électives semblent suggérer une participation traumatique.
En histologie, il existe un infiltrat histiocytaire plurifocal, formant des palissades entourant une zone de nécrose basophile souvent riche en polynucléaires neutrophiles.
Il s’y associe volontiers une leucocytoclasie avec ou sans vascularite des petits vaisseaux dermiques.
La corticothérapie générale est d’une efficacité variable.
Certains auteurs ont considéré que la dermatite interstitielle granulomateuse avec arthrite et les granulomes nécrosants extravasculaires devaient être intégrés dans un vaste groupe dénommé dermatose neutrophilique et granulomateuse palissadique liées aux complexes immuns se basant sur la présence dans ces pathologies d’une nécrose basophile.
Les différences cliniques et histologiques de ces pathologies, que les auteurs attribuent à l’évolutivité des lésions, semblent néanmoins trop importantes pour pouvoir les regrouper.
Autres causes :
A – CAUSES INFECTIEUSES :
Causes rares de GP, les lésions infectieuses granulomateuses sont parfois très nécrosantes, s’accompagnant de plages de nécrobiose riches en polynucléaires et en lymphocytes, particulièrement chez les sujets immunodéprimés (et sidéens).
Son diagnostic repose sur la mise en évidence de l’agent pathogène par des colorations spéciales (PAS, Ziehl, Gomori-Grocott) ou la culture de fragments biopsiques. Les étiologies sont bactériennes (mycobactérioses, tréponématoses, maladie des griffes du chat), mycologiques (sporotrichose, cryptococcose, coccidioïdomycose), parasitaires (schistosomiase).
B – RÉACTIONS À CORPS ÉTRANGERS :
De tels GP, bien que rares, parfois rapportés sous le terme de réactions allergiques à corps étrangers, peuvent simuler un nodule rhumatoïde ou un GA.
Leur diagnostic repose sur la notion d’intervention antérieure ou de traumatismes locaux, l’absence d’étiologie évidente, la mise en évidence inconstante d’un corps étranger (justifiant l’examen systématique en lumière polarisée de tout GP), tel que du fil de suture, du béryllium, du collagène bovin.
C – CAUSES DIVERSES :
1- Sur cicatrice de zona :
Ces lésions apparaissent à la phase de guérison de la maladie ou parfois de façon retardée et correspondent à de petites papules strictement localisées au site des anciennes vésicules, sans disposition annulaire mais dont l’histologie est celle d’un GP classique ou en rosette.
Elles pourraient être la conséquence d’une réaction d’hypersensibilité retardée secondaire à l’infestation virale.
2- Association granulome annulaire-lymphome :
Des éruptions de type GA, atypiques par leur dissémination et leur caractère douloureux, ont été rapportées au cours de lymphomes ganglionnaires (maladie de Hodgkin ou lymphome B).
Si certaines de ces associations apparaissent fortuites, d’autres semblent témoigner d’une réponse inflammatoire directement liée au lymphome, comparables aux réactions sarcoïdosiques décrites dans les lymphomes T.
3- Association granulome annulaire-immunodépression :
Les GP de l’immunodépression congénitale ou acquise sont aussi rapportés sous le terme de GA atypique.
Ils en partagent le caractère annulaire et sont souvent disséminés, parfois prurigineux correspondant histologiquement, soit à un GP classique, soit interstitiel avec rosette.
Ils sont le plus souvent observés au cours des états d’immunodépression liés au VIH.
Quelques cas ont régressé sous traitement antiviral.
Conclusion :
Les maladies et circonstances étiologiques caractérisées par un GP sont nombreuses.
Si l’examen histologique est indispensable au diagnostic, celui-ci ne peut être, dans bon nombre de cas, confirmé que par une analyse clinique très précise aidée d’une enquête à la recherche de maladies associées dont l’impact est souvent déterminant dans ce groupe d’affections.

")





")



{kind=link}