Les groupes de complémentation et le dénombrement desgènes :
L’analyse génétique d’un phénomène consiste à dénombrer le nombre de gènes
impliqués dans celui-ci, puis à identifier la fonction biochimique de chacun d’entre
eux, leurs interactions éventuelles et la régulation de leur expression.
Le dénombrement des gènes impliqués dans un phénomène est l’une des applications
du test de complémentation fonctionnelle.
En obtenant le plus grand nombre possible de mutants du phénomène étudié, on
diminue le risque de ne pas toucher l’un des gènes impliqués dans celui-ci (à moins
que les mutations d’un gène particulier soient létales), puis, en croisant entre eux
tous les mutants récessifs, on peut construire des « groupes de complémentation ».
Chaque groupe de complémentation est un ensemble de mutants ne complémentant
pas entre eux, donc touchés dans un même gène; par conséquent un groupe de complémentation définit un gène en regroupant tous les mutants (récessifs) mutés
dans ce gène.
Le nombre de groupes de complémentation définis par l’analyse fonctionnelle
des croisements entre mutants récessifs du phénomène étudié correspond
au nombre minimal de gènes impliqués dans ce phénomène.
Ce nombre de gènes peut être supérieur si on considère que le crible de mutants
n’a pas été assez efficace (aucun mutant pour certains des gènes) ou si des
mutants dominants ont été obtenus (ils ne peuvent être rattachés, par test fonctionnel,
à aucun groupe de complémentation).
La
complémentation fonctionnelle est un outil de croisement :
Croiser des drosophiles est simple, il suffit de les réunir dans un tube et de les laisser
faire, croiser des végétaux est souvent plus laborieux puisque l’expérimentateur va
devoir récupérer le pollen pour le disperser sur le stigmate de l’ovaire.
Mais comment croiser entre elles des souches d’organismes unicellulaires comme
la levure ?
En effet, l’ensemencement d’une boîte par deux souches haploïdes (de
signe sexuel opposé) va conduire à l’apparition de colonies haploïdes parentales,
là où les cellules des deux souches n’ont pas été en contact, et de colonies diploïdes
là où le contact a permis la fusion cellulaire, sans aucun moyen de distinguer les colonies
diploïdes qu’on recherche des colonies haploïdes parentales qui nous indiffèrent.
C’est ici que la complémentation fonctionnelle se révèle être un outil utile et efficace
pour l’expérimentateur.
En utilisant deux souches parentales auxotrophes pour
des molécules différentes, par exemple la valine pour l’une et le tryptophane pour
l’autre, on peut, en les croisant sur milieu minimum, être sûr de ne récupérer que des
colonies diploïdes, grâce à la complémentation fonctionnelle dont elles peuvent
bénéficier.
Les mutations d’auxotrophie des souches parentales sont ici utilisées
comme « marqueurs de sélection de diploïdes ».
Ainsi, dans une étude portant sur des mutants [his–], auxotrophes pour l’histidine,
tous les mutants [his–], comme la souche SSR [his+] doivent être porteurs de marqueurs
de sélection de diploïdes; de ce fait la souche désignée comme SSR est certes
« sauvage » pour le phénotype histidine mais obligatoirement mutée pour le (ou les)
marqueur(s) de sélection de diploïdes.
Interprétation
fonctionnelle et moléculaire de la dominance et la récessivité :
A - Approche formelle et factorielle de la dominance
et de la récessivité :
Il est d’usage de croiser un mutant avec une souche de référence, dite sauvage, afin
d’observer le phénotype du diploïde qui en est issu.
Si ce diploïde présente un phénotype
identique à celui du parent sauvage, le phénotype mutant est dit « récessif », si le phénotype est identique à celui du parent mutant, celui-ci est dit « dominant »,
enfin si le phénotype du diploïde est différent, éventuellement intermédiaire, les
phénotypes parentaux sont dits « co-dominants ».
Du fait que les phénotypes observés résultent de l’action, ou de l’inaction (en fait,
on ne sait pas), de gènes sous-jacents, l’interprétation factorielle de ces observations
phénotypiques conduit à considérer que l’allèle sauvage a, selon les cas, un effet
dominant ou récessif par rapport à l’effet de l’allèle muté, de même qu’on jugera de
l’effet dominant ou récessif d’un hétéro-allèle vis-à-vis de l’effet d’un autre hétéroallèle.
Le même type d’approche formelle et factorielle permet de considérer, en génétique
médicale que :
– une maladie génétique est dite récessive quand sa manifestation exige la présence
de deux exemplaires mutés du gène impliqué dans cette maladie.
Dans ce cas
chacun des deux parents, qui n’est en général pas atteint est dit porteur sain; il est
hétérozygote, porteur d’un exemplaire fonctionnel non muté du gène et d’un
exemplaire muté, celui qu’il a transmis à son enfant atteint, conjointement avec la
transmission de l’exemplaire muté de l’autre parent;
– une maladie génétique est dite dominante quand la présence d’un seul exemplaire
muté du gène impliqué dans la maladie suffit à sa manifestation; a fortiori quand
les deux exemplaires sont mutés la maladie est présente, mais peut, dans certains
cas, revêtir une forme plus grave, voire différente.
Dans le cas où l’individu atteint n’est porteur que d’un seul exemplaire muté du
gène (cas de loin le plus courant), cet exemplaire muté a été transmis par l’un des
deux parents, qui est lui-même atteint puisqu’il suffit d’avoir un seul exemplaire muté
pour l’être, de sorte que tout individu atteint a, en amont, l’un de ses deux parents
atteints, et, en aval, la moitié de ses enfants atteints.
Cela suppose néanmoins que la maladie dominante soit compatible avec la vie et
la reproduction, ou bien qu’elle est mortelle mais ne survient que tardivement dans
la vie (ce qui est les cas de la plupart des maladies neuro-dégénératives).
Il peut arriver cependant qu’aucun des deux parents ne soit atteint, observation qui
présente alors deux interprétations :
– soit l’enfant atteint est porteur d’une mutation de novo, apparue dans la lignée
germinale de l’un des deux parents;
– soit l’un des deux parents est porteur de la mutation mais ne présente pas la
maladie car la présence d’un allèle muté du gène n’est pathogène que sous différentes
autres conditions génétiques et/ou environnementales, conditions qui sont
réunies chez l’enfant et mais non chez le parent porteur.
On dit, dans ce cas, que
la maladie a une pénétrance incomplète, c’est-à-dire que la probabilité de manifestation,
sachant que la mutation pathogène est présente, est inférieure à 1.
Si la
présence d’un exemplaire muté suffit, quel que soit le contexte, à la manifestation
de la maladie, on dit que la maladie a une pénétrance complète.
Mais une interprétation factorielle n’est pas une interprétation fonctionnelle car
elle ne préjuge ni de l’action ni de l’inaction de l’allèle muté puisqu’on ne sait pas s’il s’agit d’une perte ou d’un gain de fonction.
Ainsi, dans l’analyse génétique formelle,
celle de Mendel ou des généticiens du début du XXe, on ne fait que « constater » la
dominance ou la récessivité des phénotypes (ou des allèles au sein d’un génotype
diploïde) sans lui apporter d’interprétation fonctionnelle puisque le gène est encore
à ce moment une boîte noire.
Avec la mise en évidence de la fonction du gène, puis le développement de la
biologie cellulaire et moléculaire, les phénomènes de dominance et de récessivité
sont devenus compréhensibles dans leur causalité fonctionnelle.
B - Les différentes mutations possibles d’un gène
et leurs conséquences fonctionnelles :
a) Distinction entre mutations de perte de fonction ou de gain de fonction
:
La modification de l’information génétique portée par un gène peut avoir deux types
antagoniques de conséquences fonctionnelles :
– soit il s’agit d’une mutation de perte de fonction, par effet quantitatif (sousexpression
conduisant à moins ou pas de produit du gène) ou par effet qualitatif
(un produit moins actif, voire inactif);
– soit il s’agit d’une mutation de gain de fonction, par effet quantitatif (sur-expression
conduisant à plus de produit du gène) ou par effet qualitatif (un produit plus actif),
ou par formation d’un produit muté doué d’une nouvelle propriété physicochimique
et biologique, qui remplace l’activité antérieure ou s’ajoute à elle (par
exemple toxicité dans certaines pathologies dominantes).
b) Distinction entre mutation ponctuelle et non ponctuelle
:
La mutation ponctuelle d’un gène est une modification locale de sa séquence d’ADN
par substitution d’une paire de base par une autre (SNP : Single Nucleotide Polymorphism)
ou par délétion ou insertion d’une, deux ou trois paires de bases. Une mutation
ponctuelle peut par conséquent conduire à une modification de l’information génétique
portée par ce gène.
Cette nouvelle version du gène constitue de toute façon, qu’elle
ait ou non une conséquence phénotypique, un allèle différent de l’allèle d’origine.
Les mutations non ponctuelles d’un gène correspondent soit à des délétions,
notamment à l’issue de mutations chromosomiques par CO (méiotique ou mitotique
ou interphasique), soit à des insertions, notamment des séquences rétrovirales ou de
type transposon, soit à l’amplification d’une courte séquence répétée, en général un
triplet.
Dans les deux premiers cas, elles conduisent principalement à des pertes de
fonction des gènes concernés, dans le troisième cas, il s’agit souvent de gain de
fonction, notamment quand le triplet répété est dans une séquence codante (maladie
de Huntington).
c) Les différents types de mutations ponctuelles du gène
:
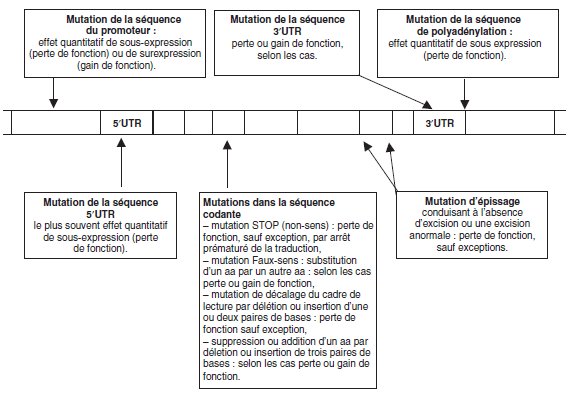
La figure 5.3, en rappelant que le gène est constitué d’un ensemble de séquences
emboîtées dont la séquence codante n’est que la séquence centrale, précise comment des mutations ponctuelles affectant diverses séquences peuvent avoir des conséquences
quantitatives ou qualitatives associées à une perte ou un gain de fonction.
On peut noter cependant que muter un gène, comme le fait de toucher un édifice
complexe, conduit le plus souvent, en probabilité, à une perte de fonction plutôt qu’à
un gain.
Bien évidemment, seules les mutations affectant la séquence codante touchent la
chaîne peptidique et se traduisent par un effet qualitatif; il faut cependant leur
adjoindre certaines mutations d’épissage qui, plutôt que de bloquer l’épissage (perte
de fonction) peuvent générer un épissage alternatif conduisant à une chaîne peptidique
anormale (avec éventuellement gain de fonction).
NB : les séquences 5′UTR (Untranslated) et 3′UTR sont les séquences transcrites,
présentes sur le messager, mais non traduites, en amont du codon AUG ou en
aval du codon STOP.
Elles ont une fonction biologique puisque la mutation de
certaines peut avoir des effets phénotypiques (dystrophie mytonique).
Figure 5.3
Remarque. Les gains de fonction sont une des bases de l’évolution génétique
sous jacente à l’évolution des espèces.
Si la fonction du gène muté est modifiée,
la fonction antérieure peut être conservée si ce gène existait préalablement
en deux copies identiques, ce qui est possible car il existe un mécanisme,
le crossing-over inégal, par lequel un gène peut se trouver dupliqué en tandem
sur un chromosome; c’est ainsi que se sont formées les familles des gènes α et β conduisant à la synthèse des différents types d’hémoglobines.
On peut
alors envisager des mutations survenant sur l’une des copies du gène, sans risque
de faire perdre la fonction du gène qui reste assurée par la copie non mutée.
d) Les différents types de mutations non ponctuelles du gène
:
➤ Les triplets répétés
Il s’agit d’une classe de mutations tout à fait particulière dont l’existence et les effets
pathologiques n’ont été observés que chez l’homme (pour l’instant).
Dans tous les
cas il s’agit d’une séquence d’un triplet répété dont la taille (le nombre de répétitions
du triplet) est variable et comprise dans une fourchette formant ainsi un polymorphisme
multi-allélique de type STR (Short Tandem Repeats) encore appelé microsatellite.
Bien que stable à la réplication, un STR peut, si sa longueur atteint ou dépasse
un certain multiple, à la suite d’une mutation ou de processus encore à l’étude,
devenir très instable sur le plan réplicatif et être alors sujet à des variations de grande
ampleur, contraction ou amplification. Dans un certain nombre de cas, l’expansion
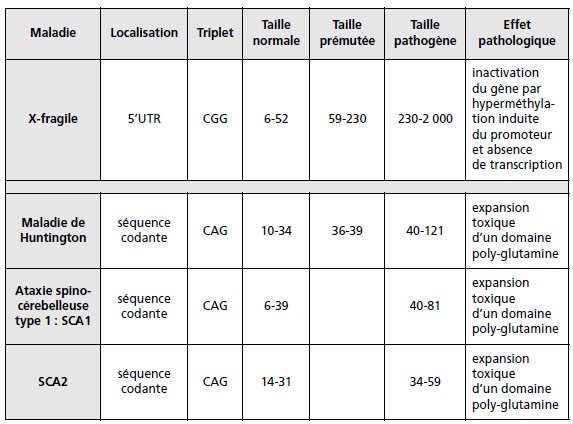
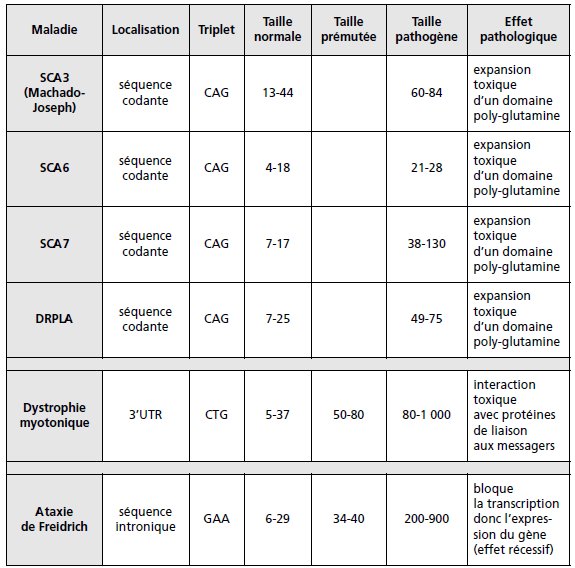
d’un triplet répété a un effet pathogène responsable des maladies à triplets (tableau 5.3).
Cet effet pathogène est dominant dans toutes les maladies dites « à triplets »
exceptée la maladie de Friedrich, le cas des maladies liées à l’X étant plus complexe,
du fait de l’interaction entre l’effet pathogène et l’inactivation de l’X, dans le sexe
féminin.
TABLEAU 5.3 LES MALADIES À TRIPLETS.
Selon les cas la séquence répétée est située dans la séquence 5’UTR, entre le
promoteur et la séquence codante (syndrome de l’X-fragile : la mutation pathogène
entraîne l’extinction du gène), dans la séquence codante (maladie de Huntington,
ataxies spino-cérebelleuses : l’expansion du CAG entraîne l’expansion toxique d’un
domaine poly-glutamine sur la protéine), dans la partie 3′ non codante de l’ARNm
(dystrophie myotonique : effet pathologique en cours d’étude), dans un intron (maladie
de Friedrich : l’expansion du GAA provoque l’extinction du gène par blocage de la
transcription par formation d’un complexe entre les quatre brins d’ADN, ce qui
explique le caractère récessif de son effet).
Ces mutations sont des mutations « dynamiques » à l’origine du phénomène d’« anticipation
génique » qui est caractérisé par la progression au cours des générations de
la gravité du phénotype (signes cliniques et âge de début des signes) en raison de
l’amplification de la séquence lors de sa transmission.
➤ Les insertions d’éléments mobiles
Les insertions d’éléments mobiles, comme les rétrovirus, peuvent inactiver un gène
ou déréguler son expression selon leur site d’insertion; c’est pourquoi certains virus
comme le HBV peuvent induire des tumeurs dans le tissu hépatique infecté.
➤ Les macromutations
Elles résultent d’accidents majeurs de la réplication ou de la recombinaison par crossingover
conduisant, selon les cas, à des déletions d’un ou plusieurs gènes, parfois à leur
duplication (crossing-over inégal), et aux diverses anomalies de structures des chromosomes
(translocations, inversions, déletions, duplication, fusions centriques).