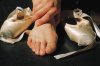
Le XP est une maladie héréditaire, grave et handicapante, se
manifestant par des altérations cutanées photo-induites souvent
associées à des lésions oculaires et parfois neurologiques.
Elle est
caractérisée par un risque très élevé de cancers cutanés, déjà
souligné dans la description initiale de l’affection par Kaposi en
1870.
La mise en évidence chez les XP, par Cleaver, d’un déficit de
réparation de l’acide désoxyribonucléique (ADN) altéré par les UV
a ouvert une voie de recherche en plein essor.
Les symptômes
s’expriment à des degrés divers d’un malade à un autre, indiquant
une hétérogénéité qui est apparue dans les études génétiques avec
l’individualisation d’une forme variante, dont la déficience est peu
comprise, et de sept groupes de complémentation déficients en
excision- resynthèse de l’ADN.
Les études récentes portant sur des
cellules isolées de ces différents groupes ont permis de cloner les
premiers gènes humains de la réparation de l’ADN et de mieux
connaître la nature et les niveaux d’intervention de leurs produits.
Les observations de XP ont, de plus, donné une grande impulsion à
l’exploration des mécanismes de la photocarcinogenèse.
Épidémiologie :
Le XP est un syndrome génétique qui atteint également les deux
sexes et qui se transmet selon le mode autosomique récessif, favorisé
par les mariages consanguins.
Ceci explique sa répartition
mondiale particulière : il est rare aux États-Unis et en Europe, avec
une incidence estimée à 1/300 000, plus fréquent au Japon
(1/100 000), mais surtout dans certains pays du Moyen-Orient et du
Maghreb, où le taux de consanguinité atteint 80 %.
L’incidence
est de 1/10 000 en Tunisie.
Le grand nombre de XP dans ces pays est également le fait de la grande taille des familles.
Le XP concerne
toutes les races, mais rarement la race noire.
Les différents groupes
de complémentation ont une fréquence respective inégale.
Étude clinique :
Les organes cibles sont la peau exposée au soleil, les yeux, et parfois
le système nerveux.
L’enfant est normal à la naissance et les
différentes manifestations sont d’apparition plus ou moins précoce
(6 mois à 3 ans) selon la gravité de la forme.
Elles s’aggravent
progressivement pour réaliser, en quelques mois ou années, un
tableau particulièrement invalidant, caractéristique du XP.
A - MANIFESTATIONS CUTANÉES :
Elles évoluent en plusieurs stades.
1- Poïkilodermie :
Un érythème des parties découvertes apparaît dès les premières
expositions au soleil, évoquant un coup de soleil, toutefois
inhabituel par son caractère persistant.
Sur ce fond, apparaissent de
multiples lentigines, brunes ou noires, et de petites taches hypochromiques, imputées à une mutation de mélanocytes sous
l’effet des UV.
Elles apparaissent sur les zones photoexposées mais
peuvent s’étendre aux régions couvertes en fonction de la sensibilité
du patient aux UV et de sa protection vestimentaire.
Des zones
atrophiques, de fines télangiectasies vont parachever le tableau de poïkilodermie des téguments insolés, si caractéristique du XP.
La
peau est fragile, elle fait le lit d’ulcérations et de surinfections
traînantes.
Elle prend, avec le temps, un aspect sec, pseudosénile,
entraînant des rétractions cutanées et muqueuses responsables
d’atrésie des lèvres, des paupières et des narines.
L’examen
histologique révèle, à un stade tardif, un aspect comparable à celui
d’une peau sénile.
2- Tumeurs cutanées
:
Leur apparition est inéluctable.
Il peut s’agir de diverses tumeurs
bénignes, de multiples kératoses actiniques et surtout de tumeurs
malignes qui impriment au XP toute sa gravité.
* Tumeurs bénignes
:
Elles sont surtout représentées par des bourgeons charnus et des kératoacanthomes, souvent multiples.
* Tumeurs malignes
:
Elles surviennent à un âge variable selon la forme de XP, dans un
éventail qui va de 2 ans jusqu’à plus de 40 ans (XP variant). Leur
nombre augmente avec l’âge, donc avec la dose cumulée d’UV
absorbés.
Il s’agit surtout de carcinomes et de mélanomes,
exceptionnellement de sarcomes.
+ Carcinomes baso- et spinocellulaires :
Ils sont largement prédominants.
Leur présentation est habituelle
mais certaines particularités méritent d’être soulignées :
– la fréquence des carcinomes baso- et spinocellulaires est, à l’âge
de 20 ans, 4 800 fois plus importante que dans la population
américaine et l’âge moyen de leur apparition est de 8 ans, soit
50 ans plus tôt que dans la population générale ;
– le siège céphalique des tumeurs est presque exclusif ;
– un même malade a successivement ou simultanément plusieurs
carcinomes baso- et/ou spinocellulaires, et le nombre atteint peut
être impressionnant (une cinquantaine) ;
– la localisation linguale du carcinome spinocellulaire est
relativement fréquente avec un siège électif à la pointe de la
langue ;
– sur le plan histologique, des images de « collision » entre
carcinome basocellulaire et mélanome peuvent être observées ;
– les métastases des carcinomes épidermoïdes sont rares.
+ Mélanomes
:
Ils sont de survenue plus tardive et plus rare que les carcinomes
mais leur fréquence est 2 000 fois plus élevée au cours du XP que
dans la population générale.
Leur fréquence est diversement
appréciée : 15 % des XP qui ont une tumeur maligne, selon une série
japonaise.
Le mélanome de Dubreuilh est le plus fréquent et
concerne avant tout les régions photoexposées.
Le mélanome peut
se révéler par une métastase d’emblée et 10 % des régressions
spontanées de mélanomes rapportées dans la littérature concernent
des XP.
+ Autres tumeurs cutanées malignes :
Elles sont exceptionnellement rapportées : fibrosarcomes, angiosarcomes, histiocytomes fibreux malins ou carcinomes sébacés.
B - MANIFESTATIONS OPHTALMOLOGIQUES :
Une atteinte oculaire bilatérale complète habituellement le tableau,
avec une gravité qui est généralement corrélée à celle de l’atteinte
cutanée. Les XP du groupe variant sont relativement épargnés.
Cette
atteinte repose sur les mêmes bases pathogéniques que les lésions
cutanées.
C’est ainsi que le segment antérieur de l’oeil, exposé aux
UV, est principalement altéré.
Les paupières sont le siège de toutes
les lésions cutanées du XP et perdent progressivement leur fonction
de protection, ce qui aggrave le pronostic oculaire.
La photophobie
est le signe le plus constant et le plus précoce, permettant d’orienter
le diagnostic dans les familles à risque, avant même les
manifestations cutanées.
Son mécanisme est inconnu. Elle confère
au malade une attitude caractéristique, tête baissée, yeux entrouverts
et larmoyants, recherchant l’obscurité.
Cette photophobie tend à
s’atténuer avec l’installation progressive d’une opacification de la
cornée.
Les conjonctives sont très altérées, hyperhémiées,
télangiectasiques, parsemées de taches pigmentaires et peuvent
s’épidermiser à la longue.
La cornée est souvent touchée par les
phénomènes inflammatoires et atrophocicatriciels.
La gravité réside
dans la survenue de tumeurs bénignes et surtout malignes
(carcinomes épidermoïdes et mélanomes), de siège palpébral,
limbique ou conjonctival, mettant en jeu le pronostic fonctionnel et
vital.
C - MANIFESTATIONS NEUROLOGIQUES :
Elles sont notées chez 14 à 40 % des XP. Leur installation est plus
tardive que les manifestations cutanées, en moyenne à 6 ans.
Leur
gravité ne semble pas proportionnelle à celle de l’atteinte cutanée.
Les différentes manifestations ne sont pas spécifiques du XP,
pouvant s’observer dans d’autres maladies hérédodégénératives.
Elles témoignent d’une atteinte centrale ou périphérique,
inégalement associées chez un même malade, et s’aggravent
progressivement avec l’âge.
Les formes les plus graves, associées à
un retard staturopondéral et à une immaturité sexuelle,
correspondent au tableau décrit par De Sanctis et Cacchione.
La
débilité mentale, le syndrome pyramidal et la neuropathie
périphérique sont les signes les plus fréquents.
1- Arriération mentale
:
Elle est notée chez 80 % des XP ayant des troubles neurologiques.
Elle est le plus souvent légère ou moyenne. Cette débilité est en
rapport avec une atrophie corticale cérébrale, objectivée par
l’examen tomodensitométrique.
Elle est aggravée par le mode de
vie des malades, non scolarisés et exclus de la vie sociale.
2- Atteinte pyramidale :
Elle se manifeste par des réflexes ostéotendineux vifs et un signe de
Babinski.
3- Neuropathie périphérique :
Il s’agit d’une neuropathie sensitivomotrice responsable d’une hyporéflexie et de troubles sensitifs.
Elle se manifeste par une
diminution de la vitesse de conduction nerveuse à l’examen électrophysiologique.
Les études histologiques ont mis en évidence
une perte importante des fibres myélinisées, avec fibrose endoneurale progressive.
4- Autres manifestations neurologiques
:
Une surdité de perception, une microcéphalie, des mouvements choréoathétosiques ou une atteinte extrapyramidale ont également
été décrits.
D - FORMES CLINIQUES :
Les différentes manifestations cutanées, ophtalmologiques et
neurologiques sont diversement associées, avec une précocité et une
gravité variables.
Cette hétérogénéité clinique correspond en fait à
une hétérogénéité génétique.
Ainsi, les sept groupes de
complémentation et le XP variant se distinguent par certaines
particularités symptomatiques et évolutives.
En l’absence
d’étude biologique, il est possible de répartir les malades selon trois
formes cliniques, dont l’intérêt pratique est surtout d’ordre
pronostique.
Pour les formes graves, la
symptomatologie est comparable à celle des malades du groupe A
et de certains du groupe G.
Le tableau des formes variantes
rejoint celui des malades du groupe F ou du XP variant.
Les formes
de gravité intermédiaire peuvent correspondre aux XP des
groupes B, C, D, E et G.
E - ASSOCIATIONS PATHOLOGIQUES :
1- À d’autres maladies de réparation de l’ADN
:
Le syndrome de Cockayne (SC) et la trichothiodystrophie (TTD) sont
deux autres maladies héréditaires, autosomiques récessives, dans
lesquelles une photosensibilité témoigne d’une anomalie de la
réparation de l’ADN par excision-resynthèse.
Le SC est caractérisé
par une dysmorphie faciale, un retard staturopondéral, des
anomalies rétiniennes et du squelette et une dégénérescence
neurologique progressive.
Un petit nombre de malades associent des
caractéristiques cliniques et biologiques du XP (groupes B, D ou G) et du SC.
Une telle association ne semble pas fortuite.
La TTD est
caractérisée par une diminution du taux d’acides aminés soufrés
dans les phanères et se manifeste par des anomalies des cheveux,
un retard mental et une petite taille.
La moitié des malades ont de
plus une photosensibilité et sont caractérisés par une mutation
génique similaire à celle du XP du groupe D, ce qui soulève
plusieurs hypothèses pathogéniques.
2- À des néoplasies internes
:
Leur fréquence au cours du XP serait 10 à 20 fois supérieure à celle
d’une population témoin. Plusieurs types de tumeurs ont été
rapportés, avec une mention particulière pour certaines tumeurs
cérébrales.
3- Autres associations :
Le caractère fortuit ou pas de certaines associations ponctuellement
rapportées se discute (lupus érythémateux, sarcoïdose ou ichtyose).
F - PRONOSTIC :
Il demeure globalement sombre et, toutes formes confondues, on
considère que les deux tiers des malades meurent avant d’atteindre
l’âge adulte.
Le pronostic vital est commandé par les cancers cutanés
et oculaires. Bien que les métastases ganglionnaires et viscérales
soient rares, les tumeurs cutanées malignes sont graves par leur
fréquence et leur multiplicité.
Elles sont à l’origine de nombreuses
complications : infections, hémorragies, anémie, cachexie ou
envahissement d’un organe de voisinage. Le décès a lieu d’autant
plus tôt que l’apparition de tumeurs a été précoce.
En fait, cette
évolution fatale est nuancée selon les différentes formes génétiques
de la maladie.
Ainsi, les XP variants ont une espérance de vie qui se
rapproche de la normale.
Par ailleurs, et à formes génétiques
comparables, la précocité et la qualité de la prise en charge peuvent
améliorer l’espérance de vie.
La fonction visuelle est compromise
à plus ou moins brève échéance, pouvant aboutir à une cécité.
L’atteinte neurologique devient également de plus en plus
invalidante.
Le préjudice esthétique est commandé par les lésions atrophocicatricielles et les séquelles des opérations chirurgicales
itératives.
Diagnostic
:
Si le malade se présente à un stade évolué, le diagnostic est facile à
poser, sur des arguments essentiellement cliniques.
Ailleurs, et en
dehors d’un contexte familial évocateur, le diagnostic est assez
difficile au début.
Les premières manifestations peuvent être
interprétées comme un simple « coup de soleil » ou comme une
toxidermie.
La répétition de l’érythème des régions photoexposées,
son caractère persistant et anormalement intense par rapport à
l’exposition solaire doivent attirer l’attention et faire envisager une
hypersensibilité anormale à la lumière solaire.
On discute alors
essentiellement les autres génophotodermatoses : le SC, la TTD, le
syndrome de Bloom ou certaines formes de porphyries héréditaires.
Chacune de ces affections a des caractères cliniques distinctifs.
Les
tests de réparation de l’ADN après exposition aux UV (unscheduled
DNA synthesis [UDS]) permettront d’apporter une orientation.
Pathogénie
:
De nombreux systèmes de réparation, bien conservés des bactéries à
l’homme, protègent la cellule contre l’accumulation de différentes
lésions d’ADN induites par l’effet génotoxique d’agents variés,
endogènes ou exogènes.
Le système de réparation par excisionresynthèse
des nucléotides ou nucleotide-excision-repair (NER) est le
plus important d’entre eux, capable d’éliminer en particulier les
altérations produites par les UV, telles que les dimères de pyrimidine
et les pyrimidines (6-4) pyrimidones, mais aussi par les radiations
ionisantes et par certains cancérigènes chimiques.
Le XP, tout comme
le SC et la TTD, répond à une anomalie dans l’une des composantes
de ce système multienzymatique et les manifestations
d’hypersensibilité au soleil observées traduisent le défaut
d’élimination des lésions photo-induites sur l’ADN cellulaire.
A - CARACTÉRISTIQUES CELLULAIRES :
Les cellules de malades XP sont particulièrement sensibles in vitro
aux rayons de 254 nm (UVC).
Cette photosensibilité cellulaire peut
être objectivée par différents tests.
1- UDS (unscheduled DNA synthesis ou synthèse
d’ADN non programmée)
:
Elle correspond à une mesure autoradiographique de l’incorporation
d’un précurseur radioactif de l’ADN (la thymidine tritiée) par des
fibroblastes préalablement irradiés aux UV.
La radioactivité sera
proportionnelle à la synthèse d’ADN réparé par excision-resynthèse,
donc faible dans les cellules de XP.
Cette méthode est utilisée
pour le diagnostic biologique et anténatal du XP.
La capacité des
cellules est exprimée en pourcentage par rapport à des cellules
témoins et les différents taux d’UDS retrouvés témoignent de
l’hétérogénéité génétique du XP.
Les patients qui ont un
phénotype XP avec une UDS normale correspondent à des XP
variants.
Leur anomalie se localise au niveau d’un autre système de
réparation, la réparation postréplicative.
2- Survie cellulaire :
Les cellules de malades XP cultivées in vitro ont une survie et une
capacité de former des colonies plus réduites que celles d’individus sains après irradiation aux UV.
L’hétérogénéité clinique du XP se
reflète à ce niveau et la gravité de la symptomatologie clinique est
inversement proportionnelle à la survie cellulaire.
Les fibroblastes
de XP variants ont une réponse normale, sauf s’ils ont été incubés
en présence de caféine, ce qui peut être utilisé comme critère de
diagnostic biologique de cette forme de XP.
3- Hypermutabilité :
Les cellules de malades XP cultivées in vitro ont un nombre plus
élevé d’anomalies chromosomiques et d’échanges entre chromatides
soeurs après irradiations aux UV que les cellules normales.
B - CARACTÉRISTIQUES GÉNÉTIQUES :
1- Groupes de complémentation :
L’hétérogénéité clinique et cellulaire du XP est l’expression d’une
hétérogénéité génétique.
En plus du XP variant, sept groupes de
complémentation (XPA, B, C, D, E, F et G) ont été individualisés
grâce à des techniques de fusion cellulaire, fondées sur la
normalisation de la synthèse réparatrice dans une cellule hybride,
issue de la fusion entre deux cellules provenant de XP de groupe
différent.
2- Gènes du XP et leurs produits
:
Les gènes impliqués dans six groupes de complémentation (XPA, B,
C, D, F et G) ont été clonés. Les complexes protéiques dépendant de
ces gènes interviennent dans plusieurs étapes du système NER.
Leur
mode d’action et l’ordre de leur intervention ne sont pas encore
définitivement établis.
Ainsi, la protéine codée par le gène défectueux dans le groupe A
montre une affinité pour l’ADN altéré et pourrait participer à la
reconnaissance de la lésion, en association probablement avec le
produit du gène impliqué dans le XPE.
Trois types de mutations ont
été identifiés au niveau du gène XPA (exon 3, 4 et 6), ce qui pourrait
rendre compte de l’hétérogénéité clinique relevée au sein de ce
groupe.
Les protéines codées par les gènes défectueux dans les
groupes XPB et XPD ont des fonctions d’hélicases, elles
interviennent pour séparer les deux brins d’ADN et démarquer la
lésion.
Il est intéressant de relever que ces deux mêmes protéines
interviennent au sein du complexe multiprotéique TFIIH qui régule
l’initiation de la transcription de l’ADN en acide ribonucléique
(ARN) messager.
Les protéines déficientes dans les groupes F et G
ont une fonction endonucléasique.
Elles font partie d’un complexe
comportant également les produits des gènes ERCC1 et ERCC11
(excision repair-cross-complementing) qui participerait à l’incision du
fragment lésé, respectivement en 5’ et en 3’.
Chez les eucaryotes, le
système NER paraît plus complexe puisqu’il possède deux voies : la
première voie est dépendante de la transcription (TCR [transcription
coupled repair]) et elle élimine les lésions au niveau de la partie
activement transcrite de l’ADN, de manière plus précoce que la
deuxième voie, celle de la réparation globale (GGR [global genome
repair]) qui intéresse le reste du génome.
Les deux voies sont
déficientes pour les différents groupes de XP, sauf pour le groupe C,
qui présente un déficit spécifique pour la GGR alors qu’il répare
efficacement les lésions situées sur le brin transcrit.
Le produit de ce
gène doit donc intervenir dans la réparation de la chromatine
inactive.
C - CARCINOGENÈSE
:
Le XP est un modèle qui illustre la présence d’une corrélation entre
l’existence de lésions d’ADN non réparées, un taux élevé de
mutations induites et une grande fréquence de tumeurs cutanées
malignes.
Le rôle du défaut de réparation de l’ADN par excisionsynthèse
a été attesté expérimentalement chez des souris mutées en
NER qui ont montré une susceptibilité accrue aux cancers cutanés.
La persistance de lésions non réparées de l’ADN engendre une
instabilité génétique et favoriserait l’apparition de mutations sur
certains gènes impliqués dans la carcinogenèse, tels que le gène p53
et les oncogènes Ha-, N- et Ki- ras.
Les mutations retrouvées au
niveau de ces oncogènes sur les cellules tumorales du XP sont de
type photo-induit, ce qui prouve bien que les cancers cutanés sont
la conséquence de l’effet génotoxique des UV.
Prise en charge :
Elle nécessite une étroite collaboration entre médecin, malade et
parents de malade et repose essentiellement sur des mesures
préventives car il n’existe actuellement aucun traitement curatif
capable de modifier l’évolution du XP.
A - PHOTOPROTECTION :
Elle doit être instaurée le plus tôt possible.
Elle peut améliorer le
pronostic cutané et ophtalmologique et retarder l’apparition de
tumeurs malignes mais ne permet pas de prévenir la dégénérescence
neurologique.
Il faut éviter l’exposition solaire entre 9 heures et
18 heures, voire mener une vie nocturne, surtout en été. Le port de
chapeau, de lunettes à filtre adapté et de vêtements couvrants est
impératif.
Des applications régulières d’un écran antisolaire, d’indice
élevé, est indispensable si le soleil ne peut pas être évité.
Cette photoprotection rigoureuse doit être compensée par une
alimentation riche en vitamine D.
B - CONSEIL GÉNÉTIQUE :
Il a pour but de déconseiller les mariages consanguins et de limiter
le nombre de naissances dans les familles à risque.
La gravité
extrême du XP et l’absence de traitement efficace justifient le recours
systématique au diagnostic anténatal (par amniocentèse ou
prélèvement trophoblastique) dans les familles à risque.
Cette
méthode ne s’applique qu’aux XP classiques.
C - TRAITEMENT DES TUMEURS CUTANÉES MALIGNES
:
Les malades et leurs parents doivent apprendre à détecter les
kératoses préépithéliomateuses.
Celles-ci pourront être détruites par
des séances périodiques de traitement électrochirurgical, par des
applications de 5-fluorouracil (crème Efudixt) ou encore par un
peeling ou une dermabrasion.
Les rétinoïdes ont également été
proposés pour prévenir l’apparition de tumeurs cutanées
malignes.
Utilisés au long cours et à forte dose, ils semblent
retarder l’apparition de kératoses actiniques mais ils sont sans effet
sur les carcinomes déjà constitués.
Pour ces derniers, le traitement
est comparable à celui qui s’applique à la population générale.
Les
tumeurs de petite taille ou superficielles peuvent être détruites, au
choix, par électrocoagulation, cryochirurgie ou laser CO2.
Pour les
autres, le traitement de première intention est l’exérèse chirurgicale.
Elle doit être la plus conservatrice possible en raison de la
multiplicité des tumeurs.
La radiothérapie n’est indiquée que dans
les cas où l’exérèse chirurgicale est impossible ou alors en
complément de celle-ci.
Elle doit être utilisée avec précaution car
une sensibilité anormale des cellules de XP aux radiations ionisantes
a été rapportée.
L’utilisation de cytostatiques par voie générale, tels
que la bléomycine, est limitée par leurs effets secondaires potentiels.
Elle est réservée aux tumeurs multiples, inaccessibles aux
traitements chirurgicaux et à la radiothérapie.
Elle peut également
être utilisée avant une exérèse chirurgicale, pour réduire la taille
tumorale. Les règles d’exérèse des mélanomes rejoignent celles qui
sont préconisées dans la population générale.
Certains auteurs ont
proposé des injections intralésionnelles d’interféron alpha.
D - PERSPECTIVES D’AVENIR :
Ces mesures préventives ont leur limite dans la prise en charge du XP
et les recherches dans le domaine thérapeutique s’orientent vers le
développement de traitements à visée étiologique.
Des essais de
correction du défaut de la réparation de l’ADN par application d’une
préparation contenant une enzyme de réparation d’origine bactérienne
sont en cours d’évaluation.
L’identification des gènes de réparation
a permis la réparation des cellules déficientes en NER in vitro, ce qui
ouvre des perspectives de thérapie génique adaptée aux XP.