Vasculites cutanées : physiopathologie, aspects anatomocliniques, classification Cours de dermatologie
Physiopathologie
:
Le développement d’une lésion vasculaire inflammatoire est dû à
des mécanismes complexes, parfois intriqués dont certains sont
encore mal connus.
1- Complexes immuns circulants
:
Le rôle des complexes immuns circulants (CIC) a longtemps été
considéré comme incontournable, selon le « très historique » modèle
de la maladie sérique.
Différents antigènes, infectieux (streptocoque, virus des hépatites B
et C, parvovirus B 19...), médicamenteux, tumoraux ou autres,
induisent la formation d’anticorps avec lesquels ils forment des
complexes antigène-anticorps.
La plupart de ceux-ci sont éliminés
par les cellules phagocytaires mais lorsqu’il existe une stimulation
antigénique répétée ou prolongée, certains complexes immuns (IC),
en excès d’antigènes, persistent.
Après s’être déposés sur les parois
vasculaires de divers organes, ils activent le complément dont
certains facteurs sont chimiotactiques pour les polynucléaires
neutrophiles.
Lorsqu’elles sont activées, ces cellules libèrent des
enzymes protéolytiques (élastases, collagénases) et des radicaux
oxygénés libres qui entraînent une nécrose pariétale.
Cette
hypothèse physiopathologique est confortée par la présence de
dépôts d’immunoglobulines (Ig) et de complément sur la paroi des
vaisseaux et d’IC circulants, parfois cryoprécipitants, dans lesquels
l’antigène responsable peut être mis en évidence. Cependant, ceux-ci
ne sont pas trouvés dans toutes les vascularites.
2- Molécules d’adhésion
:
Ces dernières années, a été mis en évidence le rôle indispensable
des molécules d’adhésion exprimées à la surface des cellules
endothéliales et des leucocytes.
De nombreuses études in vivo et in vitro impliquent surtout intercellular adhesion molecule (ICAM) 1 et vascular cell adhesion
molecule (VCAM) 1, appartenant à la superfamille des Ig, dans la
physiopathologie des vasculites, les autres molécules d’adhésion
ayant été beaucoup moins étudiées. ICAM 1, présente sur les cellules
endothéliales, se lie à l’intégrine CD11b/CD18 sur les polynucléaires
et VCAM 1 à son ligand VL4 situé sur les lymphocytes, les
monocytes et les éosinophiles.
La migration des cellules circulantes
en dehors des vaisseaux est donc favorisée par ces molécules
d’adhésion.
Leur expression est augmentée par des cytokines relarguées par ces cellules, en particulier l’interféron a, l’interleukine
(IL) 1 et le tumor necrosis factor (TNF) a qui de plus, favorisent
l’activation des polynucléaires neutrophiles et augmentent leur
adhésivité à la paroi vasculaire.
3- Immunité cellulaire :
Des phénomènes immunitaires à médiation cellulaire peuvent
également intervenir.
En effet, des cellules mononucléées
(lymphocytes T activés, monocytes, macrophages) sont souvent
trouvées au sein des granulomes périvasculaires et l’on constate
fréquemment des concentrations plasmatiques élevées du récepteur
soluble de l’IL2 et du TNFa au cours de ces vasculites.
4- Anticorps anticytoplasme des polynucléaires
neutrophiles (ANCA)
:
Récemment, les ANCA ont été impliqués dans certaines vasculites.
Mis en évidence par Van der Woude dans la granulomatose de
Wegener, ces autoanticorps sont dirigés contre des antigènes du
cytoplasme des polynucléaires neutrophiles et des monocytes.
Les
deux principaux antigènes sont la protéinase 3 (PR3), reconnue par
la plupart des c-ANCA et la myéloperoxydase (MPO) reconnue par
la majorité des p-ANCA, enzymes contenues dans les granulations
primaires de ces cellules.
Un faible pourcentage de p-ANCA
reconnaît d’autres constituants des granules primaires (élastase,
cathepsine) ou des granules secondaires (lactoferrine).
Il est possible
que ces antigènes intracellulaires puissent s’exprimer à la surface
des polynucléaires neutrophiles lorsque ceux-ci sont activés.
Détectés par immunofluorescence indirecte, on observe soit une
fluorescence cytoplasmique (c-ANCA) soit une fluorescence
périnucléaire (p-ANCA).
In vitro, ces anticorps activent les polynucléaires neutrophiles,
entraînant leur dégranulation et la production de métabolites
oxygénés, facilitent leur adhésion aux cellules endothéliales et la lyse
de celles-ci.
Les ANCA anti-PR3 diminuent aussi l’activité des
inhibiteurs des protéases (a2 macroglobuline, a1-antitrypsine).
Cependant, si certains ANCA ont probablement un rôle pathogène,
d’autres sont sans doute dus à une activation polyclonale des
lymphocytes B et sont observés dans différentes maladies
immunologiques, sans qu’ils interviennent dans leur physiopathologie.
Des anticorps anticellules endothéliales ont aussi été
trouvés dans certaines vasculites mais leur rôle est encore très flou.
5-
Autres :
D’autres mécanismes peuvent aussi être impliqués, en particulier
dans les vasculites d’origine infectieuse :
– l’antigène peut se fixer directement au niveau de la paroi
vasculaire, entraînant la formation d’IC in situ ;
– certains agents infectieux, en particulier viraux, peuvent infecter
les cellules endothéliales ;
– divers antigènes infectieux, en particulier d’origine
staphylococcique, pourraient « jouer » le rôle de superantigènes,
entraînant une activation polyclonale non spécifique des
lymphocytes T et la synthèse de cytokines pro-inflammatoires (IL1,
TNFa...).
En fait, il est probable que l’atteinte vasculaire observée dans ces
différentes affections soit due à l’intrication et/ou l’association de
plusieurs de ces mécanismes physiopathologiques.
Aspects anatomocliniques :
A -
ASPECTS HISTOLOGIQUES :
La définition des vasculites étant anatomique, le diagnostic repose
sur les données de l’anatomopathologie.
Les lésions associent à des
degrés divers un infiltrat et une nécrose de la paroi vasculaire.
L’importance et la formule de l’infiltrat sont variables.
Les
polynucléaires neutrophiles sont les principaux éléments observés
dans les vasculites leucocytoclasiques.
Le noyau de ces
polynucléaires est alors éclaté avec des débris nucléaires dispersés
dans la paroi des vaisseaux et le tissu conjonctif voisin.
Parfois
l’infiltrat est composé principalement de lymphocytes envahissant
la paroi du vaisseau atteint.
Ces véritables vasculites lymphocytaires
sont à différencier des infiltrats lymphocytaires périvasculaires
n’envahissant pas la paroi vasculaire, n’ayant pas du tout la même
signification pathologique.
En fait, les vasculites lymphocytaires
pourraient correspondre à un stade évolutif des vasculites
leucocytoclasiques marquant le passage à un stade subaigu.
La
prédominance de macrophages avec ou sans cellules géantes
caractérise les vasculites granulomateuses.
Le granulome peut se
situer dans la paroi des vaisseaux ou à distance.
Exceptionnellement,
les éosinophiles prédominent dans l’infiltrat.
La nécrose ou dégénérescence fibrinoïde de la paroi vasculaire n’est
pas toujours visible du fait d’une atteinte parfois segmentaire ou
focale comme dans la périartérite noueuse ou de la minceur de la
paroi des vaisseaux atteints associés à un infiltrat particulièrement
intense comme dans les vasculites leucocytoclasiques.
Il s’agit d’une
modification de la substance fondamentale observée au contact de
l’infiltrat notamment lorsqu’il renferme des polynucléaires
neutrophiles, à distinguer de simples dépôts de fibrine.
Diverses conséquences de l’atteinte inflammatoire peuvent être
observées telles qu’une thrombose secondaire à l’atteinte des cellules
endothéliales et à l’inflammation.
Ces vasculites thrombosantes sont
difficiles à différencier des processus initialement thrombotiques,
provoquant une nécrose colonisée secondairement par les cellules
inflammatoires.
L’intensité de la nécrose cutanée témoigne de
l’extension de l’obstruction vasculaire.
La disparition des cellules
inflammatoires peut laisser place à une sclérose mutilante angiocentrique parfois sténosante.
Une xanthomisation des lésions
est possible, notamment en présence de lésions granulomateuses ou
au cours de l’erythema elevatum diutinum.
Tous les types de vaisseaux peuvent être atteints dans la peau.
Les vasculites leucocytoclasiques touchent plus les capillaires et les
veinules postcapillaires que les artérioles du derme profond ou de
l’hypoderme.
À l’inverse les vasculites nécrosantes sont surtout
observées sur les artérioles de moyen calibre ; cependant il n’est pas
rare d’observer un aspect de nécrose fibrinoïde également dans les
vaisseaux de faible calibre au cours des vasculites leucocytoclasiques.
L’étude en immunofluorescence directe d’une biopsie cutanée en
peau lésée est d’un apport diagnostique limité.
En effet, la mise en
évidence des dépôts vasculaires d’Ig et de complément dépend de
la cause et de l’âge de la lésion.
Leur absence ne peut en aucun cas
éliminer le diagnostic de vasculite.
À l’inverse, leur présence isolée
sans lésion histologique de vasculite n’a pas de valeur diagnostique ;
ces dépôts peuvent être également présents au cours des thromboses
sans vasculite.
Cette technique d’immunofluorescence directe
peut en revanche fournir des éléments d’orientation étiologique.
Des
dépôts d’IgA sur les vaisseaux de petit calibre orientent vers le
diagnostic de purpura rhumatoïde tout en sachant que ces dépôts
ne sont ni constants, ni spécifiques au cours de cette vasculite
systémique.
Des dépôts d’IgG, IgM ou IgA et de complément
disposés en bandes granuleuses le long de la jonction
dermoépidermique sont évocateurs de lupus érythémateux tout en
n’étant pas, là encore, spécifiques, notamment en peau exposée.
Les
études comparatives d’hybridation in situ ou de biologie moléculaire
en peau atteinte et en peau saine en utilisant des marqueurs
d’antigènes de micro-organismes peuvent mettre en évidence de
façon sélective la présence de ces micro-organismes dans les
vaisseaux pathologiques.
Ces techniques, encore peu utilisées en
pratique courante, pourraient apporter une aide diagnostique,
notamment au cours des vasculites infectieuses.
B - ASPECTS CLINIQUES :
Les vasculites cutanées ont des aspects cliniques variés avec souvent
plusieurs types de lésions élémentaires associées donnant à
l’ensemble un aspect polymorphe.
Les lésions purpuriques sont infiltrées, plus ou moins nécrotiques,
localisées sur les zones déclives.
Elles sont souvent associées
à des lésions érythémateuses ou à des éléments érythématopapuleux
ou nécrotiques.
En cas de vasculites chroniques telles que celles observées au cours des cryoglobulinémies associées à une hépatite
C, le purpura est parfois masqué par une dermite ocre généralement
bilatérale non expliquée par l’insuffisance veineuse.
Les lésions papuleuses peuvent prendre des aspects différents.
Il s’agit
tantôt de lésions urticariennes particulières du fait de leur
caractère peu prurigineux et de leur persistance dans la même zone
plus de 24 heures.
Ailleurs, il s’agit de lésions papuleuses rouge
foncé pouvant prendre une disposition annulaire rappelant
l’érythème polymorphe, fréquemment localisées sur le tronc,
ne prédominant pas sur les zones déclives.
Des lésions papuleuses
sont également observées au cours de l’erythema elevatum
diutinum.
Cette rare vasculite, d’évolution chronique avec des
poussées récidivantes, est caractérisée cliniquement par la présence
de plaques, papules ou de nodules rouge sombre, violacés ou
jaunâtres, répartis de façon symétrique sur les faces d’extension des
membres et les extrémités, respectant le tronc.
Les fesses et la
région du tendon d’Achille sont aussi des sièges de prédilection.
Les nodules dermiques ou hypodermiques des vasculites sont de
petite taille, à rechercher sur les trajets vasculaires des membres,
parfois à peine visibles, toujours palpables.
Ils évoluent parfois vers
la nécrose et l’ulcération.
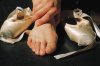
Le livedo prédomine nettement sur les membres inférieurs,
pouvant toucher également la face postérieure des membres
supérieurs et la moitié inférieure du tronc.
Il est typiquement infiltré
et ramifié lors de la phase active de la vasculite.
Il est soit isolé, soit
associé à d’autres lésions, en particulier aux nodules dermiques et
aux nécroses cutanées.
Les lésions nécrotiques sont rarement isolées, pouvant compliquer
toutes les autres lésions vues précédemment.
Elles sont parfois
superficielles à l’origine de décollements bulleux, parfois plus
profondes donnant des placards noirâtres bordés d’un liseré purpurique.
L’évolution vers une ulcération est fréquente.
Enfin une vasculite peut se manifester par une lésion pustuleuse non
folliculaire, souvent entourée d’une aréole purpurique notamment
dans la maladie de Behçet ou les entérocolites inflammatoires.
C - CORRÉLATIONS ANATOMOCLINIQUES :
À l’atteinte des vaisseaux de petit calibre du derme correspondent
les lésions purpuriques, papuleuses et pustuleuses.
À
l’opposé, les nodules traduisent souvent une atteinte des vaisseaux
plus profondément situés dans le derme ou l’hypoderme, de plus gros calibre.
Quant au livedo et aux lésions nécrotiques, elles
correspondent soit à une atteinte des vaisseaux de petit calibre du
derme, soit à une atteinte des artérioles du derme profond ou de
l’hypoderme, soit à l’association des deux.
Les vasculites
leucocytoclasiques du derme superficiel sont observées dans
toutes les vasculites cutanées isolées ou systémiques, n’orientant pas
le diagnostic étiologique.
Les vasculites nécrosantes des artérioles
de moyen calibre sont évocatrices de périartérite noueuse
tout en n’étant pas spécifiques.
En effet, des atteintes des artérioles
cutanées peuvent être observées dans les vasculites
médicamenteuses, infectieuses et dans diverses vasculites
systémiques telles que la maladie de Wegener, l’angéite de Churg et
Strauss, la maladie de Takayasu ou la polyarthrite rhumatoïde.
La nature de l’infiltrat a une valeur diagnostique en présence de
lésions granulomateuses orientant vers le diagnostic de
maladie de Wegener ou d’angéite de Churg et Strauss sans en être
spécifique.
En revanche, l’existence d’un infiltrat lymphocytaire ou leucocytoclasique a peu de valeur diagnostique.
Certains auteurs,
dans des articles anciens, ont souligné cependant la fréquence de
l’infiltrat lymphocytaire dans les vasculites médicamenteuses.
Diagnostic différentiel :
Toutes les lésions purpuriques ne correspondent pas à des
vasculites.
A - PURPURAS PAR FRAGILITÉ VASCULAIRE
:
Ils sont généralement pétéchiaux ou ecchymotiques, non infiltrés.
Cette fragilité capillaire est constitutionnelle ou acquise.
Lorsqu’elle
est constitutionnelle, elle est souvent familiale, se manifestant par
l’apparition d’un purpura localisé après des traumatismes mineurs.
Le signe du lacet est habituellement positif.
Elle peut être soit isolée,
soit s’intégrer dans des maladies plus complexes comme certains
types du syndrome d’Ehlers-Danlos.
Les purpuras acquis par fragilité vasculaire sont de diagnostic
habituellement facile du fait de leur topographie très évocatrice et
de leur contexte clinique.
– Le purpura de Bateman est formé de taches ecchymotiques de taille
variable, volontiers situées sur la face dorsale des avant-bras et des
mains, recouvertes d’une peau atrophique et ridée avec des lésions
pseudocicatricielles en étoile.
Il est observé dans le grand âge.
Son
apparition plus précoce doit faire rechercher un hypercorticisme
endogène ou iatrogène.
– La fragilité vasculaire des avitaminoses C est généralisée,
s’accompagnant d’asthénie, de douleurs musculaires, d’hémorragies
gingivales.
Le purpura cutané y est fréquemment périfolliculaire.
– Au cours de l’amylose, le purpura, en rapport avec la fragilité
capillaire liée aux dépôts périvasculaires de substance amyloïde,
prédomine dans les zones de frottement, en particulier sur les
paupières supérieures et les grands plis.
– Le syndrome des ecchymoses douloureuses ou syndrome de Gardner-Diamond apparaît plutôt chez la femme volontiers névrotique et
serait associé à une autosensibilité aux hématies ou à certains de
leurs composants.
B - ANGIODERMITES PURPURIQUES ET PIGMENTÉES
:
Elles regroupent un certain nombre de lésions dermatologiques
bénignes.
La dermite ocre de Favre et Chaix et la dermite purpurique et
lichénoïde de Gougerot-Blum sont des manifestations cutanées de
l’insuffisance veineuse chronique, localisées aux jambes.
La première
se manifeste par des macules purpuriques devenant brunâtres, ocres
avec le temps, la seconde donne des papules violines.
Elles sont
expliquées par la stase veineuse entraînant une dilatation des
capillaires du derme avec extravasation de globules rouges et dépôts
dermiques de pigments sanguins.
D’autres signes d’insuffisance
veineuse sont généralement présents : télangiectasies, nodules pseudokaposiens, remaniements sclérodermiformes...
Les autres dermites purpuriques pigmentaires sont des purpuras,
également souvent chroniques mais d’étiologie inconnue.
Leurs
aspects cliniques un peu différents leur ont valu des noms
particuliers.
Ils sont cependant très voisins histologiquement, avec
une capillarite hyperplasique, un épaississement de l’endothélium
et une péricapillarite lymphocytaire contenant des macrophages
chargés d’hémosidérine.
– La maladie de Schamberg est localisée principalement sur les
membres inférieurs.
Elle est formée de plaques mal limitées, brunes
ou rouge orangé, essaimant à la périphérie en points couleur
« poivre de Cayenne ».
– L’eczematide-like purpura, observé surtout chez l’homme, est une
dermatose purpurique et télangiectasique, débutant souvent aux
chevilles puis s’étendant vers le haut, associée à des lésions
eczématiformes prurigineuses, d’évolution spontanément régressive.
Le prurit est calmé par les dermocorticoïdes.
– Dans le lichen aureus, on observe des macules purpuriques et de
petites lésions brun orangé, parfois papuleuses, lichénoïdes, à
disposition linéaire.
– Le purpura annulaire de Majocchi apparaît à tout âge dans les deux
sexes.
Il est constitué de plaques souvent nombreuses, annulaires,
d’extension centrifuge, avec effacement central progressif donnant
une zone centrale de teinte jaunâtre et un anneau périphérique
constitué de fines télangiectasies et de points purpuriques.
La
guérison spontanée est habituelle après quelques mois ou années.
C - PURPURA THROMBOTIQUE :
– Le diagnostic différentiel entre purpura thrombotique en rapport
avec une thrombose intravasculaire et le purpura des vasculites est
parfois difficile sur la simple clinique, avant les résultats de l’examen
anatomopathologique d’une biopsie cutanée.
L’existence de placards
nécrotiques de diamètre supérieur à 1 cm, entourés d’un liseré purpurique, fait suspecter une thrombose, alors que des lésions plus
polymorphes comportant, à côté de lésions nécrotiques, des lésions
érythémateuses, du purpura infiltré ou des nodules dermiques
orientent vers une vasculite.
Cette distinction n’a pas
qu’un intérêt théorique, les purpuras thrombotiques, volontiers
extensifs, sont souvent des urgences diagnostiques ; ils relèvent de
causes différentes de celles des vasculites.
L’atrophie
blanche idiopathique ou vasculite hyalinisante segmentaire (livedoid
vasculitis des Anglo-Saxons) est un aspect clinique particulier de
thrombophilie.
La lésion primitive est purpurique, évoluant vers une
nécrose très douloureuse bordée par un réseau livedoïde.
La
cicatrisation, lente, laisse place à des lésions séquellaires,
porcelainées et atrophiques avec une bordure télangiectasique et
pigmentée.
Les lésions sont limitées aux membres inférieurs.
En histologie, il existe des thrombi hyalins des vaisseaux dermiques
sans infiltrat inflammatoire.
Le livedo est inconstant, uniquement
localisé dans les zones périulcéreuses ou plus diffus.
L’étiopathogénie de l’atrophie blanche reste obscure, probablement
non univoque du fait de la mise en évidence de facteurs variés de
trombophilie.
Certaines observations ont été intégrées dans le
syndrome des antiphospholipides.
– Les formes aiguës de coagulation intravasculaire disséminée
s’installent en quelques heures.
L’état général est rapidement altéré.
Le purpura y est volontiers ecchymotique et nécrotique.
La
dissémination des microthromboses peut être responsable de
défaillances viscérales variées : détresse respiratoire, choc
cardiogénique, insuffisance rénale ou surrénale aiguë.
Les hémorragies, conséquences de la
consommation des facteurs de la coagulation et des
plaquettes, ainsi que de la fibrinolyse secondaire, sont
d’importance variable.
Les principales causes, le diagnostic
biologique et le traitement sont détaillés ailleurs.
– Le déficit homozygote en protéine C et, plus rarement, en protéine S
est parfois à l’origine de nécroses cutanées extensives néonatales,
bordées d’un liseré purpurique réalisant un tableau clinique appelé,
à tort, purpura fulminans, terme qui devrait être réservé à la
pathologie infectieuse.
Il existe un risque d’hémorragies
intracérébrales et de thromboses viscérales multiples.
– Les déficits hétérozygotes ou acquis des protéines C ou S donnent
aussi essentiellement des lésions purpuriques et nécrotiques, parfois
précédées ou accompagnées d’un livedo, notamment chez la femme
obèse après prise d’antivitamines K.
Des lésions purpuriques
apparaissent dans les 10 premiers jours du traitement, volontiers sur
les régions fessières, les cuisses, la poitrine, s’étendant
rapidement.
Il faut arrêter momentanément l’antagoniste de la
vitamine K, supplémenter le malade en vitamine K et entreprendre
une héparinothérapie à dose efficace pour tenter de diminuer
l’étendue de la nécrose qui peut nécessiter secondairement une
greffe.
Ultérieurement, le traitement antivitamine K pourra être
repris sous contrôle strict, avec une héparinothérapie à dose
anticoagulante efficace, en utilisant une antivitamine K à long délai
d’action, administrée initialement à faibles doses et en
n’interrompant l’héparine que lorsque l’hypocoagulabilité induite
par l’antivitamine K est suffisante et stable.
– Les nécroses induites par l’héparine ou ses dérivés apparaissent
typiquement entre le cinquième et le quinzième jour du traitement,
parfois plus tôt en cas de traitement antérieur.
Elles peuvent
s’accompagner d’une thrombopénie, de saignements et de
thromboses veineuses et/ou artérielles.
Lorsque l’héparine est
administrée par voie sous-cutanée, la localisation des plaques de
nécrose aux points d’injection est un argument clinique important
en faveur du diagnostic.
Elles peuvent cependant être également
situées en dehors des zones d’injection.
L’héparine doit être
immédiatement et définitivement arrêtée avec un relais, le plus
rapide possible, par les antivitamines K.
– Les cryopathies sont à l’origine d’un purpura nécrotique, localisé
préférentiellement sur les extrémités (pieds, doigts, oreilles, nez).
Au
cours des cryoglobulinémies, la découverte d’une thrombose sans
vascularite est plus souvent observée dans les types I ou II, avec un
fort contingent monoclonal devant faire rechercher un myélome.
– Les thromboses cutanées associées aux anticorps antiphospholipides
peuvent simuler une vasculite avec des lésions purpuriques,
érythémateuses, nodulaires, ou un livedo.
Exceptionnellement, elles
se manifestent par des nécroses extensives, justifiant le recours aux
plasmaphérèses dans les formes résistantes à une anticoagulation
efficace.
– Les thrombocytémies essentielles ou secondaires à un autre
syndrome myéloprolifératif (maladie de Vaquez, leucémie myéloïde
ou splénomégalie myéloïde) sont parfois à l’origine de lésions
purpuriques et nécrotiques, généralement associées à d’autres
manifestations dermatologiques telles qu’un livedo ramifié
douloureux, symétrique et distal, un syndrome de Raynaud, une
érythrocyanose, une érythromélalgie, voire des gangrènes distales.
– Le purpura nécrotique de l’hyperparathyroïdie (primitive ou
secondaire à une insuffisance rénale chronique) évolue vers des
nécroses cutanées extensives de sombre pronostic.
Il existe une calcinose cutanée favorisant la survenue des thromboses des
vaisseaux dermiques.
– Les nécroses liées à une oxalose sont exceptionnelles.
D - PROCESSUS EMBOLIGÈNE :
Il doit être évoqué chez un polytraumatisé, en présence d’une
athérosclérose ou d’une cardiopathie emboligène, tel un myxome,
que le purpura peut révéler.
Chez le sujet polytraumatisé, les embolies graisseuses sont à l’origine
de lésions purpuriques prédominant dans les régions axillaires
antérieures ou les creux sus-claviculaires, associées à des troubles
neurologiques ou psychiatriques.
L’examen ophtalmologique révèle
fréquemment des hémorragies conjonctivales ou des dépôts
graisseux dans les vaisseaux rétiniens.
Il peut également s’y associer
une insuffisance respiratoire secondaire à un infiltrat interstitiel
diffus, une insuffisance rénale avec présence de graisses dans les
urines. Un diagnostic et un traitement précoces permettent
d’améliorer le pronostic.
Chez un sujet athéroscléreux, le diagnostic d’embolies de cristaux de
cholestérol sera évoqué devant des lésions purpuriques des membres
inférieurs associées à un livedo, des zones nécrotiques et un ou
plusieurs orteils pourpres.
Il existe généralement un syndrome
algique, les pouls sont conservés.
Des manifestations systémiques
peuvent simuler une périartérite noueuse.
Un facteur déclenchant
est volontiers retrouvé dans les semaines précédentes : geste
chirurgical vasculaire, cathétérisme artériel et/ou mise en route d’un
traitement anticoagulant (héparine, antivitamines K) ou
fibrinolytique.
La notion de cardiopathie emboligène est souvent méconnue avant
l’apparition de lésions cutanées pouvant simuler une vasculite, d’où
la nécessité au moindre doute de réaliser une échographie.
Autres manifestations
dermatologiques associées
aux vasculites systémiques
:
A - GRANULOME NÉCROSANT EXTRAVASCULAIRE
:
Initialement décrit par Churg et Strauss en 1951 comme une
manifestation de l’angéite systémique allergique, le granulome
nécrosant extravasculaire a été secondairement décrit dans diverses
vasculites systémiques et connectivites.
Il se manifeste
cliniquement par des papules ou des nodules de taille variable entre
2 mm et 2 cm de diamètre, souvent multiples, bilatéraux et
symétriques.
Leur couleur va de celle de la peau normale au rouge
violacé.
Une croûte ou une ulcération centrale est souvent présente.
Rarement, il revêt d’autres aspects cliniques trompeurs tels que des
vésicules, des pustules, des plaques arciformes ou une masse
indurée.
Les sièges de prédilection des lésions sont les faces
d’extension des coudes, des doigts, moins fréquemment les fesses,
le cuir chevelu, les faces d’extension des genoux, des mains, des
pieds, le cou, les oreilles.
Histologiquement, existe une nécrose fibrinoïde et oedémateuse du collagène entourée d’un granulome
contenant des polynucléaires éosinophiles, des histiocytes et des
lymphocytes.
Le centre du granulome contient parfois des
polynucléaires neutrophiles et des débris leucocytoclasiques.
L’infiltrat peut prendre une disposition palissadique.
Généralement,
il n’existe pas de corrélation entre l’aspect clinique, histologique et
la nature de la maladie systémique associée.
Cependant,
l’éosinophilie serait plus souvent rapportée en cas de syndrome de Churg et Strauss.
B - HYPODERMITE ET PANNICULITE :
Des lésions d’hypodermite et/ou de panniculite ont surtout été
décrites au cours de la maladie de Takayasu ainsi que dans la
vasculite observée au cours de la maladie de Behçet.
Les lésions
n’ont aucune particularité clinique ou histologique laissant présumer
le rattachement à la vasculite.
Elles siègent principalement sur les
membres inférieurs formant des nodules de plus grosse taille que
celle des nodules des vasculites nodulaires avec lesquelles elles
peuvent coexister.
C - PYODERMA GANGRENOSUM :
Des aspects de pyoderma gangrenosum ont été rarement décrits au
cours de la maladie de Wegener, de la maladie de Takayasu et de la
maladie de Behçet, généralement avec un aspect d’ulcérations
torpides.
L’histologie est non spécifique.
D - GRANULOME :
Des lésions granulomateuses sans vasculite ni nécrose centrale
peuvent être observées au cours de certaines vasculites notamment
au cours de la maladie de Wegener.
Leur aspect clinique est très
variable à type de papules, de nodules, d’infiltration sous-cutanée,
de tumeur ou d’ulcération chronique.
Tout le revêtement cutané
peut être atteint ainsi que les muqueuses adjacentes ; les localisations
les plus fréquentes étant le visage, la muqueuse buccale, les seins, le
scrotum.
Elles sont de diagnostic difficile en dehors du contexte
clinique.
E - THROMBOPHLÉBITE SUPERFICIELLE :
Tantôt de diagnostic évident du fait d’une induration douloureuse
d’un cordon veineux, tantôt de diagnostic plus difficile
essentiellement histologique devant des nodules inflammatoires, les
thrombophlébites superficielles sont surtout observées au cours de
la thromboangéite de Buerger, la maladie de Behçet et la
polychondrite atrophiante.
F - GANGRÈNES :
Les gangrènes des extrémités, résultant d’une obstruction artériolaire
ou artérielle, sont rapportées dans toutes les vasculites touchant les
artères de moyen ou de plus gros calibre.
Elles peuvent être
d’origine vasculitique, thrombotique ou embolique, ces trois
phénomènes pouvant être associés.
G - HÉMATOMES FILIFORMES SOUS-UNGUÉAUX :
Touchant souvent plusieurs doigts, des hémorragies sous-unguéales,
en flammèches, sont le plus souvent le signe d’un processus
thrombotique (syndrome des antiphospholipides) ou emboligène
(myxome cardiaque)
H - SYNDROME DE RAYNAUD :
Lorsqu’il est bilatéral, le syndrome de Raynaud atteint 5 à 30% de
la population générale.
Il est aussi classiquement associé à
toutes les vasculites.
Cependant, sa prévalence au cours de
nombreuses vasculites reste inconnue ; il a peu de valeur
diagnostique.
En revanche, un phénomène de Raynaud unilatéral
est suggestif d’une obstruction artérielle pouvant être secondaire à
une maladie de Takayasu.
Stratégie diagnostique :
Devant un aspect clinique évocateur de vasculite, la nécessité d’une
confirmation histologique du diagnostic nous paraît nécessaire étant
donné les confusions cliniques possibles avec des manifestations
thrombotiques ou emboliques et la définition histologique des
vasculites.
Une étude en immunofluorescence directe d’une lésion
est souhaitable car pouvant orienter le diagnostic étiologique.
Devant une vasculite confirmée par l’histologie, le clinicien doit
apprécier l’extension de la vasculite et en rechercher une cause.
L’interrogatoire et l’examen clinique sont d’une importance
primordiale pour apprécier l’extension de la vasculite.
En effet, ils
permettent de suspecter ou de mettre en évidence une altération de
l’état général, une atteinte articulaire (plus souvent à type
d’arthralgies que d’arthrites), une hypertension artérielle, une
atteinte digestive (douleurs abdominales, troubles du transit,
hémorragies), une atteinte musculaire (myalgies, oedèmes
segmentaires, plus rarement déficit), une atteinte du système
nerveux central ou périphérique, une atteinte oto-rhinolaryngologique
(sinusite, chute de l’audition), une atteinte oculaire
ou urogénitale.
La recherche d’une atteinte rénale, cardiaque ou
pulmonaire oblige à pratiquer systématiquement un certain nombre
d’examens complémentaires : sédiment urinaire, recherche de
protéinurie, créatinine, radiographie pulmonaire face et profil,
électrocardiogramme complété éventuellement par une échographie
cardiaque.
Il est important de répéter les examens urinaires
(bandelette urinaire et protéinurie des 24 heures) au rythme d’une
fois par semaine à une fois par mois pendant un minimum de 3
mois, voire pendant toute une année.
Le bilan étiologique doit
s’acharner à rechercher une cause.
En l’absence de toute
symptomatologie clinique orientant vers une cause, un certain
nombre d’examens complémentaires seront demandés sytématiquement : numération-formule sanguine, vitesse de
sédimentation, transaminases, cryoglobulinémie, anticorps
antinoyaux, facteur rhumatoïde, ANCA, dosage du complément
total et de ses fractions, immunoélectrophorèse des protéines
sériques, sérologies virales (hépatite B et C), anticorps
antistreptococciques.
Selon le contexte clinique, ce bilan sera complété par une sérologie du virus de l’immunodéficience humaine
(VIH), des hémocultures, une échographie cardiaque ou une
ponction lombaire.
Les autres sérologies virales (parvovirus B19,
Epstein-Barr virus, cytomégalovirus...), pour certains systématiques,
nous paraissent facultatives en raison de l’absence de conséquences
thérapeutiques à l’exception de certains cas particuliers (grossesse,
collectivités, immunodépression).
En l’absence de cause évidente, un panorex dentaire et le scanner des sinus aideront à la recherche
d’une infection focale.
Malgré ce bilan, plus de la moitié des vasculites vues en dermatologie restent de cause inconnue.
Classification :
La classification des vasculites est un casse-tête chinois auquel se
sont régulièrement heurtés depuis plus d’un siècle de grands noms
de la médecine internationale.
Au fil du temps, de nombreuses
entités ont été individualisées en raison de leurs particularités anatomocliniques, évolutives ou étiologiques.
Certaines d’entre elles
ont perduré malgré les progrès des connaissances médicales et les
vagues successives des nouvelles classifications alors que d’autres
ont été inconstamment reprises et cherchent encore leur identité.
En
fait, les classifications se heurtent à deux principaux écueils :
l’absence de corrélation entre les différents paramètres notamment
histologiques et étiologiques et la possibilité de survenue
concomitante ou successive chez un même malade de divers aspects
cliniques ou histologiques de vasculites.
Étant donné la définition
histologique des vasculites, les classifications prenant en compte la
description anatomopathologique des lésions, notamment la taille
des vaisseaux atteints, ont été régulièrement reprises.
La classification de Fauci apparaît
aujourd’hui totalement confuse et obsolète pour plusieurs raisons :
exclusion actuelle des vasculites de la granulomatose
lymphomatoïde considérée comme une hémopathie, et de
l’érythème noueux, regroupement arbitraire de diverses entités dans
le groupe des vasculites d’hypersensibilité alors que les vasculites médicamenteuses ne sont pas incluses dans ce groupe, longue liste
hétéroclite et incomplète des autres vasculites.
Les critères de
classification de vasculites de l’ACR datant de 1990 ne sont
applicables que chez des malades ayant déjà un diagnostic
histologique de vasculite ; ils ont été établis sur des malades vus en milieu
rhumatologique ; aussi sont-ils difficilement
applicables en dermatologie.
La classification
proposée par Jorizzo en 1993 est difficilement compréhensible du
fait de l’intégration dans le groupe des vasculites des nodules
rhumatoïdes, proches des granulomes extravasculaires ainsi que de
la granulomatose lymphomatoïde et des vasculites nodulaires, de
l’individualisation de la réaction lépreuse qui devrait être incluse
dans les vasculites septiques et de l’inclusion dans le groupe des
vasculites touchant les artérioles de moyen calibre de la
granulomatose de Wegener, qui intéresse préférentiellement les
vaisseaux de petit calibre.
La classification de la conférence de
consensus de Chapel Hill paraît actuellement la plus adaptée du
fait de sa simplicité et de la séparation de la périartérite noueuse
classique de la polyangéite microscopique qui ont en effet une
symptomatologie et une évolution différentes.
Cette classification
prend relativement peu en compte les causes des vasculites.
Toutes
ces remarques reflètent les limites des classifications actuelles
essentiellement issues des données anatomocliniques ainsi que notre
relative méconnaissance des aspects étiologiques et
physiopathologiques de ces affections qui se traduit par une
thérapeutique généralement empirique et aspécifique.
Aussi est-il
nécessaire d’avoir une vision pragmatique globale des vasculites et
notamment de ne pas les classer trop hâtivement sur les seules
données d’un unique examen anatomopathologique, notamment
cutané.
La classification proposée dans ce traité
privilégie les causes tout en maintenant un groupe de vasculites
représentant une entité anatomoclinique définie, généralement de
cause inconnue.
Elle est également critiquable sur différents points :
un même agent étiologique peut donner des aspects cliniques
différents ; ainsi l’hépatite B est-elle responsable de lésions urticariennes correspondant à une vasculite des petits vaisseaux
dermiques au stade prodromique et de certaines périartérites
noueuses qui pourraient également figurer dans les vasculites
infectieuses.
Au cours des vasculites associées aux hémopathies
peuvent être impliqués des agents infectieux (exemple : leucémie à
tricholeucocytes et tuberculose), des anomalies immunologiques
(cryoglobuline, hypergammaglobulinémie...) et/ou des
médicaments. Ainsi, selon le proverbe chinois, titre d’un article de
Lie :
« Plus ça change, plus c’est la même chose »