



















Introduction :
Les tumeurs cutanées conjonctives bénignes se forment à partir des éléments du tissu conjonctif propre et de ses structures différenciées telles que les vaisseaux sanguins, les muscles, les nerfs et la graisse.
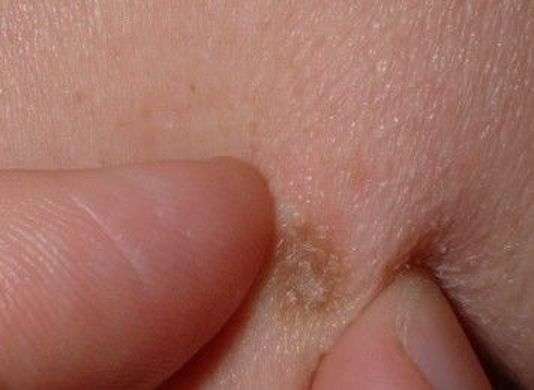 Embryologiquement, elles dérivent principalement du mésoderme.
Embryologiquement, elles dérivent principalement du mésoderme.
Elles sont fréquentes, tantôt isolées, tantôt révélatrices de syndromes systémiques.
Le diagnostic souvent uniquement suspecté cliniquement, n’est affirmé qu’après examen histologique et en ayant recours aux progrès réalisés dans les techniques immunohistochimiques.
Les tumeurs cutanées conjonctives bénignes posent de façon indiscutable un problème majeur de nosologie et de classification.
En effet, il existe un certain nombre d’états frontières où le diagnostic entre tumeur bénigne et maligne n’est pas aisé.
De même, la distinction entre tumeurs vraies et pseudotumeurs réactionnelles ou inflammatoires n’est pas toujours facile.
En revanche, les ostéomes, tumeurs dérivées de la cellule souche mésenchymateuse, seront intégrés dans cette étude, bien que le tissu osseux ne soit pas un constituant normal de la peau.
Tumeurs fibreuses :
A – DERMATOFIBROME (DF) :
Fibroma simplex, histiocytome cutané, histiocytome fibreux, histiocytofibrome (HCF), fibrome en « pastille », fibrome dur, DF lenticulaire, fibrose nodulaire sous-épidermique, hémangiome sclérosant…
Cette abondante synonymie est due aux nombreuses formes histologiques et clinicopathologiques de DF posant de multiples problèmes de diagnostic différentiel.
On décrit un large spectre de tumeurs dont l’histologie va du fibrome pur (composé de façon exclusive ou presque de fibroblastes et de faisceaux de collagène) à l’histiocytome pur (riche en cellules phagocytaires à l’aspect d’histiocytes), en passant par l’HCF.
1- Clinique :
Le DF apparaît chez l’adulte avec une prédilection pour le sexe féminin.
C’est une formation papuleuse ou nodulaire intradermique, ferme, parfois ombiliquée, unique ou multiple, de quelques millimètres de diamètre à 2 ou 3 cm au maximum.
L’épiderme fait corps avec la tumeur.
Il est responsable de la couleur de la lésion allant du rose au noir foncé, en passant par le rouge et le brun chamois qui sont les teintes les plus habituelles.
Sa surface est lisse ou kératosique.
Il est indolore mais peut devenir prurigineux ou douloureux. L’ulcération est exceptionnelle.
La compression latérale de la tumeur entraîne une dépression centrale en forme de fossette assez caractéristique.
Il existe des DF aplatis, de niveau avec la peau avoisinante (fibrome en pastille de Civatte), voire même déprimés. Mais, dans les deux cas, la palpation retrouve l’infiltration dermique.
La topographie est ubiquitaire, mais avec une préférence pour les membres inférieurs.
Dans un petit nombre de cas, on retrouve la notion d’un traumatisme antérieur (piqûre d’insecte, plaie superficielle, cicatrice vaccinale) avec un temps de précession variable.
La tumeur est soit de croissance lente suivie d’une période de stabilisation sur plusieurs années, soit de croissance rapide.
La régression spontanée est suivie d’une cicatrice hyperpigmentée légèrement atrophique.
Le traitement est chirurgical.
Les récidives sont le fait des exérèses incomplètes et sont rares (< 1 % des cas).
* Formes cliniques :
– Les DF multiples disséminés peuvent survenir au cours de lupus systémique, de myasthénie, de syndrome de Sjögren, de pemphigus vulgaire traités par des immunosuppresseurs.
Des DF multiples éruptifs ont également été décrits chez des sujets infectés par le virus de l’immunodéficience humaine (VIH).
Les lésions simulent le plus souvent un sarcome de Kaposi, mais toutes les lésions papuleuses de nature infectieuse ou non infectieuse susceptibles d’être rencontrées chez le sujet VIH doivent être envisagées dans le diagnostic différentiel.
– Les HCF multiples groupés (ou fibrome extensif en « nappe ») correspondent à de multiples éléments papulonodulaires érythématopigmentés, très fermes, faisant saillie sur un large placard assez bien limité, infiltré, parfois linéaire.
L’évolution très lentement progressive sur des années est marquée par la multiplication des éléments nodulaires.
Les lésions siègent surtout aux membres inférieurs, dans la région lombaire et de la ceinture pelvienne.
– L’HCF avec phénomène de Meyerson est exceptionnel.
Il se traduit par l’apparition d’un halo érythématosquameux, asymptomatique, séparé de la lésion primitive par quelques millimètres de peau saine donnant un aspect en « cocarde ».
Ce halo correspond en histologie à une image d’eczéma.
L’exérèse de l’HCF entraîne la disparition du halo.
– L’HCF avec épithélioma basocellulaire est une association qui fut longtemps contestée et qui reste exceptionnelle.
La modification clinique de l’HCF doit faire évoquer la survenue d’un épithélioma (apparition d’un nodule, troubles pigmentaires bleutés ou noirâtres, bordure perlée, ulcération).
2- Histologie :
Il s’agit d’une prolifération intradermique, non encapsulée, bien délimitée par rapport aux structures avoisinantes, séparée de façon inconstante de l’épiderme par une bande de collagène indemne (grenz zone) et atteignant rarement l’hypoderme qui la délimite en deçà.
La tumeur est constituée, en proportions variables, de cellules fusiformes fibroblastiques et de cellules arrondies ou ovalaires histiocytaires volontiers enchevêtrées entre elles selon une structure tourbillonnante.
Dans les formes à prédominance fibreuse, les cellules fusiformes ressemblent à des fibroblastes avec un noyau allongé et un cytoplasme réduit.
Les cellules sont entourées par des fibres de collagène fines ou organisées en d’épais faisceaux.
C’est en périphérie que la sclérose collagénique est la plus marquée et que les cellules infiltrent les faisceaux de collagène hyalinisé.
Les cellules fusiformes adoptent fréquemment, du moins focalement, une disposition storiforme.
Dans le type cellulaire (« histiocytome »), de nombreuses cellules ont un aspect d’histiocytes. Les colorations spéciales révèlent des dépôts d’hémosidérine ou de lipides.
Les cellules parfois volumineuses, géantes, plurinucléées sont du type cellules géantes à corps étrangers ou présentent un cytoplasme abondant, riche en graisses, lui donnant un aspect spumeux (histiocytoxanthome ou xanthohistiocytome ou histiocytome xanthomisé).
Dans les deux types, il faut noter :
– des dilatations vasculaires avec de petites hémorragies, mais les dépôts d’hémosidérine sont normalement ténus ;
– un infiltrat inflammatoire lymphohistiocytaire à la périphérie de la tumeur ;
– des modifications très fréquentes de l’épiderme sus-jacent à la tumeur avec, le plus souvent, une hyperplasie et une acanthose.
De façon plus rare, mais non exceptionnelle, un aspect de kératose séborrhéique ou un aspect pseudoépithéliomateux est rencontré.
Il peut exister également une hyperplasie basaloïde simulant un épithélioma basocellulaire superficiel. Une hyperpigmentation de la couche basale est souvent présente.
Enfin, l’épiderme d’épaisseur normale peut s’accompagner d’une disparition de la papillomatose dermique.
Il semble que plus les tumeurs fibrohistiocytaires sont haut situées dans le derme, plus les modifications épidermiques sont importantes.
La rareté des mitoses ainsi que des atypies, la différenciation cellulaire, l’absence de pléomorphisme sont des éléments importants du diagnostic de même que le mode d’extension en profondeur de la tumeur, les rapports architecturaux entre la tumeur et l’hypoderme, les modifications de l’épiderme en regard.
* Formes histologiques du DF :
Au fil des années, nous assistons à une multiplication dans la description de formes histologiques de DF, parfois associées à des particularités cliniques. Nous ne mentionnerons que les principales ou les plus récentes.
Zelger et al ont proposé de classer les DF en quatre groupes en fonction de leurs particularités : architecturales ; cellulaires ou stromales ; architecturales et cellulaires, ou stromales ; le quatrième groupe correspond à des formes mixtes au sein d’une même tumeur.
+ Formes avec particularités architecturales :
– HCF angiomatoïde (HFA).
L’HFA décrit par Santa-Cruz en 1981 est une forme rare touchant le tronc et les membres de la femme d’âge moyen et se caractérisant par un nodule de petite taille, parfois douloureux, rouge foncé ou marron bleuté.
Sa caractéristique histologique est la richesse en cavités vasculaires remplies de sang, de taille variable allant d’une simple fente à une vaste cavité pseudokystique.
Des globules rouges extravasés sont dispersés dans la tumeur.
Les dépôts d’hémosidérine intra- et extravasculaires sont ici très importants par rapport à la forme classique.
– HCF cutané palissadique.
Cette tumeur s’individualise par l’arrangement nucléaire palissadique autour d’une zone centrale de collagène hyalinisé, pouvant toucher 10 à 70 % de la lésion.
Cet aspect est surtout central, tandis qu’en périphérie c’est l’image d’un HCF classique.
– DF atrophique.
La présentation initiale est celle d’un nodule ou, plus souvent, d’une lésion aplatie, rétractée, donnant l’impression clinique d’un épithélioma basocellulaire sclérodermiforme ou d’une cicatrice.
Ce sont plus volontiers les épaules et les membres supérieurs qui sont atteints.
La particularité histologique est la dépression centrale et l’amincissement très net du derme.
Il n’y a pas d’atrophie épidermique mais, au contraire, les modifications classiques d’un HCF.
Les fibroblastes et le stroma collagénique sont en proportions variables.
De façon constante, existe une pénétration de la graisse sous-cutanée.
– HCF sous-cutané et profond.
La tumeur solitaire, souscutanée affecte l’adulte entre 30 et 50 ans et les deux sexes sans véritable préférence.
Siégeant surtout aux membres inférieurs, elle peut être ubiquitaire dans sa topographie (tête, cou, tronc).
Outre le tissu sous-cutané, elle peut avoir des localisations profondes dans les muscles et les séreuses (mésentère).
Après exérèse, les récidives sont fréquentes (20 % des cas).
L’histologie montre une tumeur très bien circonscrite, de grande taille (plusieurs centimètres), entourée d’une pseudocapsule dense et fibreuse, à la différence de l’HCF classique.
Le monomorphisme lésionnel est également une particularité, ainsi que l’importance de l’architecture storiforme.
Les mitoses sont présentes mais n’excèdent pas cinq mitoses/champ X 10.
Cette tumeur est à différencier de l’HCF malin (HFM) pléomorphe, du dermatofibrosarcome protuberans, vu l’importance de l’architecture storiforme et de l’hémangiopéricytome par l’existence possible de vaisseaux multipes branchés entre eux et de diamètre luminal variable.
+ Formes avec particularités cellulaires ou stromales :
– Histiocytome à cellules épithélioïdes.
Cette entité décrite en 1989 par Wilson-Jones se présente comme une papule ou un nodule ferme, sessile ou pédiculé, simulant un granulome pyogénique, de 0,5 à 1,5 cm, siégeant surtout au niveau des membres inférieurs d’une femme adulte d’âge moyen.
La lésion, bien délimitée, constitue une masse ovoïde du derme papillaire en surplomb par rapport à la peau avoisinante, délimitée à sa base par une collerette épidermique.
Il existe une certaine uniformité cytologique puisque 50 à 80 % des cellules tumorales sont de larges cellules épithélioïdes, cuboïdes, triangulaires, anguleuses, au cytoplasme éosinophile abondant.
L’arrangement cellulaire se fait en « mosaïque » au sein d’une matrice finement collagénique.
Mêlées aux cellules épithélioïdes se trouvent, en nombre variable, cellules fusiformes et histiocytaires. Le réseau vasculaire est très marqué, avec parfois un aspect d’hémangiopéricytome.
C’est avec un nævus de Spitz que le diagnostic différentiel devra être fait en priorité.
– DF à différenciation myofibroblastique. La différenciation myofibroblastique peut être présente dans un DF (10 à 20 % des cas) mais en proportion faible (< 25 % des cellules tumorales).
En revanche, Zelger et al ont mis en évidence de façon rare dans le DF la présence d’un contingent myofibroblastique important, excédant 25 % et pouvant atteindre 90 % des cellules tumorales.
Les myofibroblastes sont identifiés par l’immunohistochimie et la microscopie électronique.
– DF à cellules granuleuses.
De nombreuses tumeurs bénignes et malignes en dehors de la tumeur d’Abrikossoff sont susceptibles de présenter en leur sein des cellules granuleuses.
Le DF en fait partie avec une proportion parfois significative de cellules à cytoplasme éosinophile granuleux.
Les granules sont PAS (periodic acid Schiff) positifs et diastase-résistants.
La réactivité des cellules granuleuses pour les marqueurs des histiocytes/macrophages donne des résultats divergents, négatifs pour Soyer et al et positifs pour Zelger et al.
La microscopie électronique montre dans le cytoplasme la présence de nombreuses vacuoles remplies d’un matériel granuleux ainsi que des structures électron-denses suggestives de phagolysosomes.
– DF à cellules claires.
Ce type rare de DF a pour originalité la présence d’un infiltrat important (> 90 %) de cellules ovalaires ou polygonales, aux bords nets, au cytoplasme optiquement clair, et dont le noyau arrondi ou ovalaire est volumineux, hyperchromatique, renfermant un nucléole unique, éosinophile et très net.
Des cellules granuleuses peuvent se mêler à ces cellules claires.
Le diagnostic différentiel se pose avec une métastase cutanée d’un carcinome rénal, un xanthome papuleux, un xanthogranulome, un sarcome à cellules claires.
– DF chéloïdien. Tout aussi exceptionnel, le DF chéloïdien décrit chez des sujets asiatiques présente une zone circonscrite pseudochéloïdienne faite de fibres épaissies, éosinophiles de collagène orientées irrégulièrement dans la partie superficielle d’un DF par ailleurs typique.
Dans cette zone, les fibres élastiques sont absentes.
– Autres. On décrit par ailleurs des formes atypiques pseudosarcomateuses, à cellules monstrueuses, sclérosantes, hémosidériniques ou sidérifères, myxoïdes, à cellules pseudoostéoclastiques avec ou sans ossification.
+ Formes avec particularités architecturales et cellulaires ou stromales :
– Histiocytome fibreux cellulaire bénin.
Cette tumeur solitaire des sujets adultes jeunes ou d’âge moyen affecte les membres comme un HCF classique.
Mais, à la différence, elle touche plus volontiers les hommes, est de plus grande taille, siège surtout aux membres supérieurs et dans des sites rares pour un HCF, comme la face et les régions acrales.
Les récurrences après exérèse sont fréquentes (dans près de 25 % des cas), quelquefois multiples mais non destructrices.
Le diagnostic histologique est difficile.
Ce sont des lésions très cellularisées, faites de cellules surtout fusiformes éosinophiles à noyau effilé.
Le polymorphisme cellulaire est limité mais, par endroits, on constate la présence de cellules musculaires lisses.
De façon constante, les cellules tumorales s’étendent au derme réticulaire profond et, dans près d’un tiers des cas, à la graisse souscutanée superficielle.
L’architecture de la tumeur est surtout fasciculaire.
L’activité mitotique d’importance variable (1 à 10 mitoses/champ X 10) est constante.
Par places, existent des foyers de nécrose. Le diagnostic différentiel doit se faire avec le léiomyosarcome et le dermatofibrosarcome protuberans.
– DF avec prolifération musculaire lisse.
La présence de cellules musculaires lisses est possible dans un DF avec surtout une prolifération de celles-ci situées en périphérie du DF.
Les cellules sont marquées de façon intense par la desmine et l’actine musculaire spécifique. Le diagnostic différentiel est celui du dermatomyofibrome.
– Angiohistiocytome à cellules multinucléées.
Il survient plus chez des femmes que chez des hommes, âgées en moyenne de 60 ans.
Les lésions multiples, parfois confluentes, sont bien circonscrites, fermes, arrondies, en forme de dôme, à surface lisse et de couleur rouge-brun ou violacée.
Elles sont localisées en majorité aux membres inférieurs et sur la face dorsale des mains et des doigts.
L’histologie montre une prolifération vasculaire de capillaires et de veinules, avec des cellules endothéliales faisant protrusion dans la lumière et des cellules plurinucléées éparses dans la lésion.
Quant aux fibres de collagène, elles sont épaissies.
L’évolution peut se faire vers la régression spontanée.
* Immunomarquage :
Les cellules tumorales ont pour constante d’être positives pour la vimentine.
Le marquage par le facteur XIIIa donne des résultats variables, liés à l’interprétation qui est faite de toute positivité constatée.
Il importe de différencier celle des cellules tumorales de celle des cellules dendritiques dermiques.
Les DF présentent souvent un contingent de cellules facteur XIIIa positives qui reste faible et localisé à la périphérie de la tumeur.
Cependant, dans certains cas, le contingent de cellules positives est tellement important que des auteurs comme Cerio et al ont vu, dans le dendrocyte, la cellule originelle de la prolifération dermique.
Le dermatofibrosarcome protuberans peut, lui aussi, présenter des cellules dendritiques marquées par le facteur XIIIa, mais très éparses et en nombre réduit.
Le CD34 a permis de progresser dans l’étude des tumeurs fusiformes dermiques et dans le diagnostic différentiel du DF avec le dermatofibrosarcome protuberans.
Le CD34 marque 50 à 100 % des cellules fusiformes du dermatofibrosarcome protuberans.
Cependant, à peu près 10 % échappent à un tel marqueur.
Les DF peuvent, quant à eux, dans quelques cas, être positifs pour le CD34.
La positivité est en général focale, confinée à la périphérie de la tumeur, avec un marquage de moins de 20 % de la totalité des cellules.
Cependant, dans des cas plus exceptionnels, la positivité est forte et touche 30 à 60 % du contingent tumoral.
Les marqueurs macrophagiques (Ki-M1P, KP1, Mac 387) donnent des résultats très aléatoires allant de l’absence de marquage à des marquages très fortement positifs.
La présence de quelques cellules positives pour l’actine musculaire lisse n’est pas rare et fait évoquer la présence de myofibroblastes.
Les marquages positifs avec l’á-1 antitrypsine et l’á-1 antichymotrypsine sont non spécifiques.
* Microscopie électronique :
D’intérêt plus dogmatique que pratique, elle identifie un spectre de cellules avec deux pôles et des aspects transitionnels entre eux.
À un pôle, on trouve des cellules d’apparence fibroblastique, fusiformes, avec un noyau unique ovalaire, un réticulum endoplasmique rugueux (RER) et un appareil de Golgi bien développés.
Dans quelques cas, l’aspect est celui de myofibroblastes, au noyau allongé, avec nucléole proéminent, riches en RER et en appareil de Golgi, dont le cytoplasme renferme de nombreux filaments intermédiaires, et présentant quelques plaques d’attachement et, par endroits, des zones de lame basale.
À l’autre pôle, les cellules sont arrondies, d’aspect histiocytaire.
La richesse en mitochondries et en phagolysosomes contraste avec le moindre développement du RER et du système golgien.
3- Diagnostic différentiel :
L’HCF peut être confondu avec d’autres lésions bénignes de la peau comme le neurofibrome (NF), le léiomyome, voire la fasciite nodulaire (FN).
C’est l’HCF dans ses multiples variantes histologiques qui pose le plus de problèmes de diagnostic différentiel.
Ceux-ci ont été envisagés et discutés dans les chapitres correspondants.
Cependant, c’est surtout dans l’HCF sous-cutané et profond, ainsi que dans l’histiocytome fibreux cellulaire bénin que le diagnostic différentiel avec une tumeur maligne, comme le dermatofibrosarcome protuberans et l’histiocytome fibreux malin, se révèle le plus difficile.
C’est l’analyse des données cytologiques et architecturales telles que nous les avons définies dans le DF qui peuvent orienter le diagnostic.
Dans le dermatofibrosarcome protuberans, la prolifération située dans le derme et le tissu sous-cutané est monomorphe, très dense, adoptant une architecture storiforme et infiltrative, marquée par l’absence de cellules géantes, de cellules xanthomisées et le faible infiltrat inflammatoire.
De plus, l’épiderme en regard est le plus souvent aminci.
Dans l’histiocytome fibreux malin, la tumeur est profondément située, marquée par un pléomorphisme important, des mitoses atypiques, des foyers de nécrose et d’hémorragies.
Les immunomarquages, et en particulier le marquage par le CD34, sont parfois utiles, mais l’insuffisance des marqueurs est à prendre en compte.
Les progrès de l’immunohistochimie n’ont donc pas permis de résoudre tous les problèmes diagnostiques.
C’est la confrontation des données cliniques à celles de l’histo-immunologie, la répétition des biopsies, l’examen de la totalité de la pièce d’exérèse qui permettront d’établir un diagnostic.
4- Histogenèse :
L’histogenèse des tumeurs fibrohistiocytaires n’est toujours pas élucidée, que l’on considère le DF comme une tumeur vraie ou comme un processus réactionnel.
Les progrès réalisés en immunohistochimie n’ont pas permis la résolution de ce problème, d’autant qu’il importe de différencier le marquage des cellules tumorales et celui des cellules réactionnelles avant de se livrer à toute interprétation hâtive.
B – TUMEUR FIBROHISTIOCYTAIRE PLEXIFORME :
Décrite par Enzinger et Zhang, cette tumeur présente à la fois les caractères de l’HCF et des fibromatoses, mais en diffère par une structure multinodulaire ou plexiforme.
1- Clinique :
À prédominance féminine, de survenue fréquente chez le sujet jeune, elle atteint, dans près de 90 % des cas, les sujets de moins de 30 ans.
La tumeur solitaire est profondément située dans le derme et le tissu sous-cutané, mal circonscrite et de consistance ferme ou caoutchouteuse.
Ce sont les membres supérieurs et les épaules qui sont les plus touchés.
De croissance lente et insidieuse, les récurrences, après exérèse, sont très fréquentes, localisées et souvent précoces.
Les métastases sont exceptionnelles et seuls deux cas de métastases ganglionnaires survenues après récidive locale sont rapportés.
Cependant, une exérèse chirurgicale large est souhaitable.
2- Histologie :
Elle montre une tumeur, de 1 à 3 cmou plus, mal délimitée, siégeant dans le derme inférieur mais pouvant s’étendre au tissu souscutané, voire aux muscles.
La composante cellulaire est variable selon la tumeur.
Trois types sont possibles.
La forme fibrohistiocytaire comprend de petits îlots ou amas de cellules à l’aspect d’histiocytes et de fibroblastes, mêlés à des cellules géantes.
Dans le type fibroblastique, les cellules fusiformes s’entrelacent en faisceaux selon une structure plexiforme.
Enfin, certaines tumeurs allient les caractéristiques des deux premiers. Le nombre de mitoses est faible, n’excédant pas deux/champ X 10.
Les immunomarquages sont positifs pour la vimentine et pour certaines cellules avec l’actine musculaire lisse.
La microscopie électronique démontre le polymorphisme cellulaire avec des cellules mésenchymateuses primitives sans caractères spécifiques, des fibroblastes actifs et des myofibroblastes, mais aussi des cellules géantes plurinucléées et des cellules xanthomateuses.
Il n’existe pas de différenciation histiocytaire évidente.
Cette tumeur, de diagnostic difficile, peut être confondue avce un très grand nombre de tumeurs bénignes et malignes des parties molles dont les principales sont l’HCF, la tumeur à cellules géantes, l’hamartome fibreux, le NF plexiforme, le neurothécome, la tumeur desmoïde, le fibrosarcome et l’HFM.
Son profil évolutif, le risque de survenue de métastases font que sa place dans le cadre des DF comme le préconisent Zelger et al nous apparaît discutable et expliquent que nous l’ayons traitée dans un paragraphe à part.
C – DERMATOMYOFIBROME :
Cette entité a été décrite par Kamino et al en 1992 pour définir une prolifération cutanée bénigne sous forme de plaque de fibroblastes et de myofibroblastes.
1- Clinique :
Classiquement, le dermatomyofibrome se présente sous la forme d’une lésion en plaque dermique, ferme, circonscrite, de couleur brun-rouge, asymptomatique, solitaire, siégeant à la partie supérieure du tronc et en particulier à l’épaule et à la région axillaire.
Ce sont surtout les femmes et les adultes jeunes qui sont atteints.
La lésion mesure 1 à 2 cm dans son plus grand diamètre.
Cette tumeur peut cependant se présenter sous la forme d’un nodule, adopter une topographie linéaire et s’accompagner de microsatellites, être de grande taille, siéger dans d’autres sites comme la paroi abdominale antérieure, le cou, la cuisse.
2- Histologie :
La tumeur, non encapsulée, bien circonscrite, est faite d’une prolifération uniforme de cellules fusiformes séparées par de fines fibres de collagène et arrangées en faisceaux entrelacés et orientés parallèlement à la surface de la peau, réalisant un aspect de plaque dans le derme réticulaire.
Les cellules tumorales sont fusiformes avec un cytoplasme éosinophile, un noyau allongé et étiré renfermant un ou deux nucléoles et une chromatine fine.
Le nombre de mitoses est faible (< deux mitoses/champ X 10) et il n’existe pas d’atypies cellulaires.
Les annexes sont épargnées. Les vaisseaux sanguins sont augmentés en nombre.
Les colorations spéciales comme le trichrome de Masson démontrent le caractère fin des fibres de collagène séparant les cellules entre elles, et l’existence d’épais faisceaux de collagène entre les faiceaux de cellules fusiformes tandis que l’orcéine montre des fibres élastiques présentes, voire augmentées en nombre, et épaissies par rapport au derme voisin.
Les immunomarquages objectivent un marquage positif des cellules pour la vimentine et l’actine musculaire, et plus variable avec l’actine musculaire lisse.
La desmine, le CD34, la protéine S100, le facteur XIIIa sont négatifs.
La microscopie électronique révèle la présence d’un double contingent cellulaire avec des fibroblastes et des myofibroblastes.
Le diagnostic différentiel peut se poser cliniquement et histologiquement avec un dermatofibrosarcome protuberans en plaque, un DF et une chéloïde.
3- Traitement :
L’exérèse n’est pas suivie de récidives.
D – FIBROME MOU (FM) :
(Molluscum pendulum, acrochordon, skin-tag.)
1- Clinique :
C’est une tumeur molle, pédiculée, couleur chair, souvent petite, filiforme, parfois volumineuse mais avec une base d’implantation mince, unique ou multiple, siégeant surtout au niveau des paupières, du cou, des aisselles, de la région sous-mammaire, de la partie supérieure du thorax.
Asymptomatique, le FM devient sensible quand il se tord et s’infarcit sur son pédicule d’implantation.
Ce sont souvent les sujets obèses qui présentent des FM.
Le FM est à différencier d’un nævus pédiculé ou d’un NF.
2- Histologie :
Dans le derme, le collagène est densifié ou parfois lâche, associé à des vaisseaux sanguins dilatés.
Au niveau de l’axe conjonctif, les annexes sont absentes.
L’épiderme en regard est aplati ou hyperkératosique et acanthosique.
Une pigmentation de la couche basale est parfois présente.
3- Traitement :
Il repose sur la cryothérapie et l’électrocoagulation.
E – FIBROME PÉRIFOLLICULAIRE (FPF) :
Le FPF est une tumeur rare, prenant naissance dans la gaine conjonctive du follicule pileux.
1- Clinique :
Le FPF se traduit par l’apparition d’une papule de quelques millimètres de couleur chair, blanchâtre, parfois rosée, ferme, à surface lisse, parfois centrée par un poil ou d’aspect pseudocomédonien.
Solitaire, le FPF siège surtout au nez, au front, au tronc, à la commissure labiale.
Multiples, les FPF atteignent la face (front, joues), le cou, la partie supérieure du tronc et des bras. Ils apparaissent entre la quatrième et la sixième décennie de vie. Ils sont parfois familiaux.
Ils peuvent s’observer dans le syndrome de Birt-Hogg-Dubé dont on connaît l’association à une polypose colique, et ce à côté des fibrofolliculomes, des FM et des trichodiscomes.
Il est souhaitable, en cas de FPF multiples, de rechercher une polypose colique.
2- Histologie :
Le FPF correspond, dans le derme réticulaire supérieur, à une densification concentrique du collagène autour d’un follicule pileux non altéré, nettement délimité par rapport au derme voisin et quelquefois par de véritables fentes.
Les fibroblastes sont au sein de la tumeur en nombre accru, surtout en cas de FPF solitaire.
Le diagnostic différentiel est parfois difficile avec le fibrofolliculome, tumeur impliquant l’épithélium folliculaire.
3- Traitement :
On préférera, en cas de lésion solitaire, la chirurgie et, en cas de lésions multiples, l’électrocoagulation.
Les récidives sont possibles.
F – ANGIOFIBROME (AF) :
Les AF constituent des tumeurs fibrovasculaires solitaires ou s’intégrant dans le cadre d’une phacomatose.
Il existe de nombreuses parentés anatomopathologiques entre les mal nommés adénomes sébacés de type Pringle de la sclérose tubéreuse de Bourneville (STB), les tumeurs de Koënen, la papule fibreuse du nez, les papules perlées de la couronne du gland (ou leur équivalent vulvaire) et même le fibrokératome digital acquis.
Ackerman et Kornberg proposent d’intégrer ces différentes lésions dans un même cadre dénommé acroangiofibrome.
1- Clinique :
L’AF est une lésion papuleuse, ferme, en forme de dôme, de petite taille (< 5 mm de diamètre), à surface lisse, parfois télangiectasique, de couleur chair ou rose-rouge.
L’AF solitaire siège au niveau du nez (arête nasale ou ailes), réalisant la papule fibreuse du nez.
Les lésions multiples siègent au niveau du visage dans la région médiofaciale.
Elles sont présentes par dizaines ou par centaines, quelquefois regroupées en de vastes masses tumorales particulièrement inesthétiques.
Plus rarement, elles sont localisées de façon unilatérale, dans un même territoire anatomique.
Elles s’intègrent le plus souvent dans un tableau clinique évocateur de STB, les AF y étant présents dans près de 90 % des cas.
Les AF apparaissent entre l’âge de 5 et 10 ans, avec une poussée évolutive à la puberté.
Mais on sait actuellement que les AF multiples peuvent s’observer au cours de la neurofibromatose (NF) de type 2, du syndrome des néoplasies endocriniennes multiples (MEN 1).
Dans le MEN 1, les AF sont de plus petite taille et moins nombreux que dans la STB, et touchent des zones normalement épargnées dans celle-ci comme les lèvres.
Le fibrome périunguéal (tumeur de Koënen) constitue une variante topographique d’AF.
Les lésions dans la STB y sont multiples, touchent plusieurs sites anatomiques et en particulier les orteils.
Le fibrome périunguéal peut cependant être unique et affecter un seul ongle, d’un orteil le plus souvent.
2- Histologie :
L’AF de siège dermique s’individualise par la présence d’une prolifération vasculaire, de volumineuses cellules fibroblastiques, étoilées (glial-like cells) et d’une fibrose collagénique, les fibres élastiques étant absentes.
La microscopie électronique identifie des cellules d’allure fibroblastique.
Les immunomarquages de peu d’intérêt et d’interprétation difficile ont surtout mis en évidence des cellules stromales dermiques marquées par le facteur XIIIa ou le CD34.
3- Histogenèse :
L’histogenèse de même que la nature des AF (tumeur vraie ou processus réactionnel) restent mal connues.
Un début de réponse semble possible avec la mise en évidence, chez des patients atteints de MEN 1, d’une délétion allélique du gène au sein même de l’AF, non retrouvée dans les AF de la STB.
Ainsi, des lésions identiques pourraient être la conséquence de délétions différentes de gènes suppresseurs de tumeurs, impliqués dans des entités différentes.
4- Traitement :
La lésion unique fera l’objet d’une exérèse chirurgicale ou d’un traitement par laser.
Dans les lésions multiples, de nombreux essais thérapeutiques ont été faits : curetage, cryochirurgie, peeling, dermabrasion, shaving-dermabrasion et enfin laser.
Plusieurs types de laser ont été essayés.
Le laser CO2 semble plus performant que le laser argon en cas de lésions volumineuses et multiples.
Le laser à colorant pulsé a pour principal intérêt d’éclaircir des lésions très congestives.
Il peut constituer un appoint et un complément thérapeutique. Le recours aux lasers a un intérêt cosmétique évident.
Vu le profil évolutif des AF, il importe de ne pas traiter trop précocement.
G – FIBROKÉRATOME DIGITAL ACQUIS (FKA) :
Le FKA ou fibrokératome acral est une entité anatomoclinique isolée en 1968 par Bart, d’étiologie inconnue, et où l’hypothèse traumatique est suggérée par la topographie acrale, bien qu’inconstante.
1- Clinique :
Dans la forme typique, cette tumeur acquise touche surtout l’homme entre 30 et 60 ans.
Elle est le plus souvent unique, mais des lésions doubles sont possibles.
La localisation aux doigts est la plus fréquente mais on connaît des FKA sur les orteils, la plante des pieds, la paume et le dos des mains, le poignet, le genou, la cheville et le mollet.
Sur les doigts, le FKA est à proximité des interlignes articulaires, sur la face dorsomédiane ou latérale des doigts.
Le FKA est un nodule indolore, saillant, ferme, de quelques millimètres à 1,5 cm, hémisphérique, de couleur peau normale, à surface plus ou moins kératosique simulant quelquefois une corne cutanée.
Sa base est séparée par un sillon de la collerette épidermique hyperkératosique dont le FKA semble émerger.
Il récidive peu après exérèse.
* Autres formes cliniques :
Ce peut être l’aspect d’un doigt surnuméraire ou d’une lésion en dôme simplement surélevée par rapport à la peau voisine.
Quant au fibrome en « gousse d’ail » ou garlic-clove fibroma, il s’agit d’une variante topographique du FKA, localisée au niveau de la matrice ou d’une rainure latérale de l’ongle, parfois à la place de l’ongle et, le plus souvent, au premier orteil.
Il y simule un botryomycome, une tumeur de Koënen, mais le diagnostic est plus nosologique qu’histologique.
2- Histologie :
Kint et al individualisent trois types anatomopathologiques sans corrélation anatomoclinique.
Le type I, le plus fréquent, correspond à la description de Bart d’une tumeur fibroépithéliale, constituée d’un axe conjonctivovasculaire recouvert par un épithélium hyperkératosique et acanthosique.
Les faisceaux de collagène épais, denses, étroitement serrés entre eux et disposés parallèlement à l’axe vertical, sont en continuité avec le tissu conjonctif normal sousjacent.
Ils sont mêlés à des fibroblastes, à des fibres élastiques amincies et à un riche réseau vasculaire.
Le type II s’individualise par la profusion des fibroblastes regroupés en faisceaux.
Le type III diffère des précédents par la paucicellularité, l’importance des phénomènes oedémateux, la pauvreté en vaisseaux et en fibres de collagène irrégulièrement disposées, et la présence possible d’un matériel mucoïde.
3- Traitement :
Il est chirurgical.
H – ÉLASTOFIBROME (EF) :
Cette lésion bénigne fibroélastique rare a été décrite, en 1959, par Jarvi et Saxen.
1- Clinique :
L’EF touche de préférence les femmes et les sujets âgés (sixième et septième décennies).
La tumeur, de croissance lente, est souvent peu ou pas symptomatique (douleur, sensibilité, gêne à la mobilisation).
Elle peut être de découverte fortuite lors d’une intervention chirurgicale pour une autre cause ou lors d’une autopsie.
Elle atteint souvent une taille importante (3 à 10 cm).
Elle est ferme, profondément située, adhérente aux structures sous-jacentes mais non à la peau.
Elle peut exceptionnellement l’ulcérer. Son siège électif est la région périscapulaire (sous- ou infrascapulaire).
Elle est uni- ou bilatérale avec souvent une lésion controlatérale de petite taille, d’apparition asynchrone ou passée inaperçue.
Des topographies inhabituelles sont possibles : région sous-olécrânienne, deltoïdienne, partie latérale du thorax, sein, grand trochanter, tubérosité ischiatique, doigt, pied et même estomac, grand épiploon, conjonctive.
Les examens complémentaires (échographie, scanner, résonance magnétique nucléaire) n’apportent qu’un diagnostic de présomption.
Il est impératif, en cas de forte suspicion d’EF, de réaliser un examen bilatéral, la mise en évidence d’une masse controlatérale augmentant la probabilité diagnostique.
Ces examens objectivent une tumeur des parties molles, dense, fibreuse, mêlée à des zones adipeuses.
2- Histologie :
L’EF est une masse ferme, non encapsulée, aux limites imprécises, adhérente aux plans profonds.
Elle est constituée d’une trame conjonctive, dense, fibreuse, peu cellularisée, assez peu vascularisée, englobant des îlots graisseux.
La trame fibreuse est faite d’un mélange de faisceaux de collagène et de fibres élastiques.
Celles-ci, après colorations spéciales, ont des aspects très particuliers : fibres épaissies, denses, plus ou moins sinueuses et ramifiées, de contours irréguliers, spiculées en « dents de scie » ; fibres fragmentées avec étranglements successifs donnant un aspect perlé ; petites masses globuleuses grossièrement arrondies comme détachées des structures précédentes.
La composante cellulaire, rare, est représentée par des fibroblastes sans atypies nucléaires.
En microscopie électronique, le polymorphisme des fibres élastiques est confirmé.
Une configuration en forme de fleur est notée, avec un axe central, dense, entouré par des spicules radiaires d’un matériel granuleux et filamenteux d’électrondensité variable.
Les cellules de l’EF correspondent à des fibroblastes et, pour certaines, à des myofibroblastes.
Les fibres élastiques sont en connection étroite avec les fibroblastes.
3- Histogenèse :
L’EF n’est pas considéré comme une véritable tumeur mais comme une pseudotumeur secondaire à la friction mécanique de l’épaule sur les côtes et aux microtraumatismes répétés dus à la sollicitation excessive de l’articulation de l’épaule, par exemple chez des travailleurs manuels ou du côté de la main prédominante.
Il s’agirait plus d’une élastodysplasie que d’une dégénérescence du tissu collagène.
L’élastogenèse apparaît perturbée avec un arrangement anormal des microfibrilles d’élastine.
Pour Kumaratilake et al, les cellules périostées seraient à l’origine des anomalies de la fibrillogenèse élastique.
Cependant, l’existence de formes familiales suggère une prédisposition génétique avec un déficit enzymatique sous-jacent.
4- Traitement :
Il est chirurgical, suivi de récurrences en cas d’exérèse incomplète.
Aucun cas de transformation maligne n’a été rapporté.
I – FASCIITE NODULAIRE (FN) :
La FN a été décrite pour la première fois sous l’appellation de fibromatose (fasciite) sous-cutanée pseudosarcomateuse, dénomination justifiée par son aspect histologique inquiétant.
Il s’agit cependant d’une prolifération fibroblastique bénigne, profonde, volontiers développée aux dépens de l’aponévrose, pouvant s’étendre à la graisse sous-cutanée et infiltrer le muscle sous-jacent.
La FN se caractérise par sa croissance rapide, sa richesse cellulaire et son importante activité mitotique.
Par certains aspects, elle ressemble à un processus néoplasique et pose donc un problème de diagnostic différentiel avec les sarcomes.
C’est une tumeur de l’adulte jeune, très fréquente.
1- Clinique :
La FN touche en priorité l’adulte de 20-35 ans.
Aux âges extrêmes de la vie, la FN est plus rare. Il n’existe aucune prédominance de sexe.
La présentation clinique est univoque, sous forme d’un nodule solitaire, profond, bien limité, ferme, asymptomatique, de petite taille en général inférieure à 3 cm, atteignant parfois des dimensions extrêmes de 10 cm.
La lésion est de croissance rapide et donc d’apparition brutale.
La phase préopératoire n’excède pas 1 mois, dans près d’un cas sur deux.
La FN est ubiquitaire dans sa topographie. Sa localisation élective est représentée par les membres supérieurs, bras et surtout avant-bras.
Par ordre de fréquence décroissant, on enregistre ensuite l’atteinte du tronc, de la tête et du cou (surtout chez l’enfant), plus rarement des membres inférieurs, des régions acrales des membres, enfin plus exceptionnellement de la glande mammaire et de la vulve.
Au niveau de la sphère oropharyngée, les localisations profondes sont possibles et à ne pas méconnaître.
2- Histologie :
La tumeur nodulaire est plus ou moins bien circonscrite, mais non encapsulée, parfois siège de cavités pseudokystiques sur la tranche de section.
Les cellules tumorales ont un aspect de fibroblastes immatures. Les mitoses sont fréquentes mais sans atypies.
Les cellules forment des faisceaux entrelacés au sein d’une trame collagénique et réticulinique, et d’une matrice mucopolysaccharidique alcianophile, disparaissant après action de l’hyaluronidase.
La substance fondamentale peut, par son importance, donner à la FN un aspect franchement « myxoïde » et faire discuter un HFM dans sa variété myxoïde, voire un myxome.
Dans les espaces extracellulaires, les fentes pseudovasculaires sont quasi constantes, la lumière des capillaires étant parfois oblitérée par la turgescence des cellules endothéliales.
L’infiltrat inflammatoire est le plus souvent réduit, comprenant quelques lymphocytes et cellules géantes plurinucléées.
Les processus d’ossifications et de calcifications sont exceptionnels.
Il est classique de distinguer trois types de FN : le type sous-cutané fait de nodules arrondis ou ovalaires, bien circonscrits, sous-cutanés et attachés au fascia superficiel, type de loin le plus fréquent ; le type intramusculaire plus ovoïde et plus volumineux ; le type intrafascial moins bien circonscrit, dont l’activité mitotique est plus importante, proliférant le long du fascia, ayant tendance à infiltrer les septa fibreux interlobulaires de la graisse sous-cutanée.
Ces variantes topographiques présentent peu de différences dans leurs caractéristiques cytologiques et architecturales.
La microscopie électronique identifie un double contingent cellulaire, fibroblastique et myofibroblastique.
Le diagnostic différentiel peut se poser avec les fibromatoses, et en particulier avec le myofibrome dans sa forme solitaire ainsi qu’avec une tumeur desmoïde.
C’est cependant un fibrosarcome qui devra être éliminé en priorité.
La densité et le monomorphisme cellulaires, l’activité mitotique intense, les multiples atypies, la paucité du matériel collagénique, mucoïde et des éléments inflammatoires, la taille de la lésion, sa localisation profonde, constituent des éléments importants du diagnostic, souvent associés à une histoire clinique de phase préopératoire plus longue.
3- Formes anatomocliniques :
Quelques variantes anatomocliniques de la FN ont été rapportées.
La fasciite crânienne prend naissance dans le périoste.
Elle touche l’enfant de moins de 2 ans, est d’apparition brutale, siège plus volontiers au niveau de l’os temporopariétal qu’au niveau frontal ou occipital.
La lésion solitaire atteint la table externe de l’os, mais quelquefois l’extension se fait jusqu’à la table interne ou à la duremère sous-jacente.
La radiographie du crâne permet d’identifier une lyse osseuse souvent associée à un anneau sclérotique périphérique.
En histologie, on trouve un aspect classique de FN, mais s’associe volontiers une formation osseuse réactionnelle avec des spicules d’os mature.
Ces tumeurs ne sont pas plus agressives que la FN classique.
La fasciite intravasculaire se caractérise par une prolifération intravasculaire affectant un ou plusieurs vaisseaux, artère ou veine, de petit et moyen calibre, avec souvent une participation de l’intima, de la média et de l’adventice.
Elle suggère souvent une tumeur maligne avec infiltration vasculaire.
La fasciite dermique siège dans le derme, l’épiderme en regard étant normal, séparé de la lésion par une bande claire, ou bien atrophique, ulcéré avec une collerette épithéliale de délimitation.
Ces tumeurs très rares peuvent infiltrer le tissu sous-cutané.
4- Pathogénie :
L’étiologie de la FN reste inconnue.
L’hypothèse d’une prolifération fibroblastique déclenchée par un traumatisme local ou par un processus inflammatoire non spécifique localisé a été suggérée.
Cependant, la notion d’un traumatisme antérieur n’est souvent pas retrouvée.
5- Traitement :
Il est chirurgical.
Les récidives sont rares, dans 1 à 2% des cas, et souvent le fait d’exérèses incomplètes.
Les formes récurrentes obligent à une relecture attentive des lames histologiques afin de ne pas méconnaître le diagnostic de fibrosarcome.
J – FIBROME DES GAINES NERVEUSES :
Le fibrome des gaines nerveuses a été décrit en 1979 par Chung et Enzinger.
1- Clinique :
Cette tumeur de croissance lente se présente comme un nodule souscutané, solitaire, ferme, peu ou pas douloureux, adhérent aux gaines tendineuses et siégeant en priorité à la main (paume, doigts), au poignet et au pied. Sa taille excède rarement 2 cm.
Il est exceptionnellement ulcéré.
2- Histologie :
La tumeur, bien délimitée, localisée dans le derme superficiel et le tissu sous-cutané superficiel est plurilobulée.
Les lobules sont souvent délimités par des espaces à type de fentes.
Chaque nodule est constitué de faisceaux de collagène dense, hyalinisé, entrelacés, mêlés à une population plus ou moins éparse de cellules fibroblastiques fusiformes ou étoilées.
Par places, des connexions étroites sont observées avec les structures tendineuses
Des zones de transition existent entre des plages de collagène hyalinisé peu cellularisées et des zones plus cellularisées. Les mitoses, jamais atypiques, sont plus fréquentes dans ces zones de forte cellularité.
Une autre caractéristique de cette tumeur est la présence de fentes vasculaires, parfois très marquées, avec un arrangement concentrique du collagène autour des vaisseaux. Des foyers de matériel mucoïde sont parfois rencontrés.
La tumeur présente volontiers dans le tissu conjonctif adjacent des expansions pseudopodiques ainsi que de petites formations nodulaires.
Le diagnostic différentiel se pose avec la tumeur à cellules géantes des gaines nerveuses, mais le fibrome des gaines nerveuses est moins cellularisé et dépourvu de cellules géantes plurinucléées et de cellules xanthomateuses.
Les tumeurs très cellularisées peuvent être confondues avec une FN, un DF, voire un fibrosarcome.
La microscopie électronique montre la présence de cellules à la fois fibroblastiques et myofibroblastiques.
3- Pathogénie :
La pathogénie du fibrome des gaines nerveuses n’est pas claire, processus tumoral vrai ou processus fibrosant réactionnel.
Récemment, Dal Cin et al ont mis en évidence des anomalies chromosomiques de type clonal au sein de la tumeur avec translocation t (2 ; 11) (q31 – 32 ; q12) faisant suggérer une origine tumorale et non réactionnelle de la prolifération. ¦ Traitement Il est chirurgical.
Les récidives semblent fréquentes (25 % des cas), surtout liées à la présence des expansions dans le tissu conjonctif adjacent.
4- Tumeurs musculaires :
Les léiomyomes (L) cutanés sont des tumeurs bénignes relativement rares, développées aux dépens des cellules musculaires lisses.
Il est classique de distinguer :
– les L pilaires ou piloléiomyomes prenant naissance au niveau des muscles arrecteurs des poils, uniques ou multiples ;
– les L génitaux prenant leur origine dans les fibres musculaires lisses du dartos scrotal ou des grandes lèvres ainsi que dans le muscle aréolaire ;
– les angioléiomyomes qui sont des L développés à partir des fibres musculaires lisses des parois vasculaires.
K – PILOLÉIOMYOMES OU LÉIOMYOMES PILAIRES :
Ce sont les plus fréquents des léiomyomes.
1- Clinique :
Les L pilaires sont des tumeurs à type de papules ou de nodules, de couleur chair ou rosée ou rouge-brun, voire rouge-violet.
Ils sont fermes à la palpation, mobiles sur les plans profonds et une de leurs particularités est la douleur spontanée ou déclenchée par le froid, la pression, un traumatisme ou des facteurs de stress.
La douleur très caractéristique n’est pas l’apanage de cette tumeur et doit faire discuter d’autres tumeurs douloureuses comme un neurome, une tumeur glomique, un spiradénome eccrine, un angiolipome.
Certaines lésions peuvent, en se contractant, s’accompagner de signes généraux (nausées, vomissements, défécation, perte d’urines, dilatation pupillaire, hypotension, pâleur).
Les L surviennent sans préférence de sexe, à n’importe quel âge, mais plutôt chez l’adulte jeune.
Ils peuvent être uniques et siéger alors plus volontiers au niveau des membres.
Les L multiples ont une topographie ubiquitaire avec une répartition souvent à parts égales entre les membres et le tronc.
Certains L adoptent une répartition linéaire ou ont tendance à se regrouper dans une zone localisée du tégument.
Le visage, les faces latérales du cou sont également des localisations possibles, bien que moins fréquentes.
Les lésions multiples apparaissent de façon sporadique ou s’intègrent dans un contexte familial à transmission autosomique dominante et à pénétrance variable.
Leur nombre peut atteindre quelquefois la centaine et leur taille, de quelques millimètres ou de l’ordre du centimètre, est inférieure à celle observée dans les lésions solitaires.
Les L multiples seraient légèrement plus fréquents que les L solitaires.
Il n’existe pas de tendance à la régression spontanée. Au fil du temps, les lésions préexistantes augmentent de taille, tandis que de nouveaux éléments apparaissent et souvent par poussées.
C’est dans les formes familiales et multiples que l’on a décrit chez la femme l’association possible à des myomes utérins.
Cette léiomyomatose cutanéo-utérine s’accompagne parfois d’une polyglobulie par augmentation de l’activité érythropoïétique au sein du tissu tumoral.
2- Histologie :
Les L sont des tumeurs localisées dans le derme réticulaire, s’étendant parfois au derme papillaire ou plus ou moins en profondeur vers le tissu sous-cutané, non encapsulées, à l’architecture nodulaire, surtout lorsqu’il s’agit de lésions multiples.
Ils sont constitués de faisceaux entrelacés de cellules fusiformes, éosinophiles, dont le noyau en forme de cigare sur les coupes longitudinales est tout à fait caractéristique.
Les coupes transversales montrent un noyau central, arrondi, avec un halo périnucléaire.
Les filaments intracytoplasmiques sont colorés en rouge foncé par le trichrome de Masson et en pourpre par l’hématoxyline phosphotungstique acide.
Les cellules ne présentent pas d’atypies et il n’y a pratiquement pas de mitoses observables lors d’une analyse champ/champ.
Enfin, il n’existe pas de phénomène de nécrose.
Raj et al ont mis l’accent sur des anomalies relativement fréquentes de l’épiderme en regard de la tumeur avec une hyperplasie, une acanthose et une papillomatose.
Le diagnostic différentiel du léiomyome solitaire se pose parfois pour l’histologiste avec un léiomyosarcome très bien différencié.
Les critères de malignité étant souvent mal définis dans les tumeurs musculaires lisses, ce sont les lésions avec une très faible activité mitotique ou un pléomorphisme très focal et très discret qui poseront le plus de problèmes diagnostiques.
Les immunomarquages montrent la positivité constante des cellules tumorales pour la desmine, l’actine musculaire lisse.
Les marquages par la protéine S100, l’énolase neurospécifique, les neurofilaments mettent en évidence, lorsqu’ils sont réalisés, une augmentation des petites fibres nerveuses dans le stroma présent au sein de la tumeur et dans celui situé dans son voisinage immédiat.
En microscopie électronique, les cellules sont typiques de cellules musculaires lisses, avec des myofilaments, des vésicules de pinocytose et des structures de type lame basale.
3- Traitement :
Il dépend du caractère symptomatique ou pas de la tumeur.
La forme solitaire relève de l’exérèse chirurgicale.
Les récurrences sont fréquentes. Les formes multiples constituent un défit thérapeutique.
Les phénomènes douloureux sont mal expliqués : contraction des fibres musculaires lisses, rôle des fibres nerveuses localisées au sein de la tumeur, compression nerveuse pour les tumeurs les plus volumineuses.
Les lésions les plus invalidantes feront l’objet d’une chirurgie.
Les diverses drogues proposées à titre antalgique, comme la nifédipine ont souvent des effets décevants.
L – LÉIOMYOMES GÉNITAUX :
On regroupe sous cette appellation les L du sein, du scrotum et des grandes lèvres.
Les L génitaux sont des tumeurs solitaires, rares, survenant à n’importe quel âge.
La lésion est bien délimitée, ferme, profonde, plus souvent asymptomatique que douloureuse.
Les L du mamelon (aréole) se rapprochent, par leur taille et leur histologie, des piloléiomyomes.
Les L de la vulve et du scrotum sont des tumeurs souvent plus volumineuses et très bien circonscrites.
Au niveau vulvaire, les L présentent des modifications myxoïdes et une hyalinisation fréquentes.
M – ANGIOLÉIOMYOME OU LÉIOMYOME VASCULAIRE :
C’est une tumeur sous-cutanée, rarement intracutanée, de croissance lente, arrondie, solitaire, n’excédant pas 4 cm de diamètre, siégeant avec prédilection sur les membres inférieurs de femmes d’âge mûr (quatrième-sixième décennies de vie).
La topographie est cependant ubiquitaire (tronc, face, cavité buccale, région sous-unguéale).
La douleur est inconstante et ce d’autant plus qu’il s’agit de formes faciale et tronculaire.
L’angio-L se distingue des précédents par sa nette délimitation et son caractère encapsulé.
Il est constitué de faisceaux de fibres musculaires lisses à disposition concentrique autour de vaisseaux nombreux, dont la lumière est de taille et de forme variables, arrondie ou parfois réduite à l’état de fentes.
Les vaisseaux dépourvus de limitante élastique interne sont plus évocateurs de veines que d’artères.
Il existe, par places, des zones de dégénérescence mucoïde, en particulier dans les volumineux angio-L.
Les cellules tumorales prennent parfois un aspect épithélioïde tout en gardant les marqueurs immunohistochimiques des cellules musculaires lisses (angioléiomyome épithélioïde).
L’angio-L montre une positivité des cellules tumorales pour les marqueurs des cellules musculaires lisses et une positivité des cellules endothéliales pour le facteur VIII antigénique, le CD31 et le CD34.
L’angio-L peut poser un problème de diagnostic différentiel avec les tumeurs glomiques s’individualisant par la présence de filets nerveux, mais surtout avec l’angio-lipo-léio-myome ou angiomyolipome.
Cette tumeur se caractérise par la présence de trois composantes (vaisseaux, cellules musculaires lisses et tissu adipeux) dont la proportion est variable.
Le tissu adipeux constitué d’adipocytes matures fait partie intégrante de la prolifération et les cellules graisseuses peuvent infiltrer la paroi des vaisseaux.
Le traitement chirurgical n’est, dans la règle, pas suivi de récidives.
Tumeurs vasculaires :
Les tumeurs vasculaires posent de façon indiscutable un problème de classification.
Il n’en existe aucune qui soit véritablement satisfaisante, d’autant que ces dernières années ont été l’occasion de voir émerger un très grand nombre d’entités différentes.
Pour cette raison, nous proposerons une classification largement inspirée des travaux récents de Requena et Sangueza.
Les malformations vasculaires ne sont pas traitées dans ce chapitre, de même que les états dits « hyperplasiques », à l’exception du granulome pyogénique, lésion très fréquente, source d’erreurs diagnostiques, et l’hyperplasie endothéliale papillaire intravasculaire, particularité histologique.
A – GRANULOME PYOGÉNIQUE (BOTRIOMYCOME) :
Le granulome pyogénique correspond plus à un tissu de granulation avec composante vasculaire prédominante qu’à une véritable tumeur.
1- Clinique :
Cette lésion très fréquente touche aussi bien la peau que les muqueuses, et la muqueuse buccale en particulier.
Elle revêt l’aspect d’un nodule rouge vif, rouge-brun, bleu-noir de taille variable (quelques millimètres, voire plusieurs centimètres), pédiculé ou sessile, asymptomatique.
Sa survenue après un traumatisme même minime n’est pas rare.
Il s’agit d’une lésion exophytique à croissance rapide, saignant facilement et présentant une consistance friable.
L’épiderme de surface est soit lisse et atrophique, soit ulcéré et verruqueux. Le granulome pyogénique survient de préférence chez les enfants et les adultes jeunes.
Les lésions sont uniques ou multiples, regroupées dans un même territoire ou au contraire disséminées, avec un caractère éruptif.
Elles peuvent prendre cet aspect éruptif sous l’influence de facteurs hormonaux comme la grossesse ou la prise de contraceptifs oraux ainsi que lors de traitement par rétinoïdes.
Dans ces cas, les lésions ont tendance à rétrocéder spontanément en quelques mois.
Enfin, une forme clinique particulière correspond à un granulome pyogénique récurrent entouré de microsatellites multiples, non pédiculés, d’apparition rapide et siégeant dans un territoire localisé, en particulier au tronc et à la région scapulaire.
2- Histologie :
Les lésions précoces sont constituées de lobules riches en veinules et en petits capillaires, au sein d’un infiltrat inflammatoire polymorphe, d’un stroma riche en mucine et pauvre en collagène.
Les cellules endothéliales sont hyperplasiques.
À l’état initial, l’aspect est celui d’un tissu de granulation, tandis qu’à un stade tardif, c’est la fibrose qui prédomine.
Les immunomarquages montrent une positivité des cellules endothéliales pour le facteur VIII et la lectine Ulex europeus.
3- Traitement :
Le granulome pyogénique pouvant régresser spontanément, le traitement ne sera en aucun cas agressif.
Il répond à la destruction de la tumeur par électrocoagulation ou par exérèse chirurgicale afin de disposer d’une étude histologique en cas de doute diagnostique, avec un sarcome de Kaposi par exemple.
B – HYPERPLASIE ENDOTHÉLIALE PAPILLAIRE INTRAVASCULAIRE :
Décrite à l’origine par Masson sous l’appellation d’hémangioendothéliome végétant intravasculaire, l’hyperplasie endothéliale papillaire intravasculaire ne constitue pas une entité spécifique.
Elle apparaît plus comme un processus réactionnel hyperplasique des cellules endothéliales, en réponse à une thrombose vasculaire avec une organisation secondaire du thrombus que comme une tumeur vraie.
Elle se développe plus dans les veines que dans les artères. Elle survient de novo dans un vaisseau dilaté ou se greffe sur une lésion vasculaire préexistante.
1- Clinique :
La clinique est celle de la lésion dans laquelle se développe l’hyperplasie papillaire.
Il peut s’agir d’hémangiome, de granulome pyogénique, d’angiokératome, d’anévrismes capillaires, de lymphangiomes, d’hématomes, de phlébectasies….
Quand la lésion semble apparaître de novo, elle réalise une papule ou un nodule pourpre ou violine, localisé le plus souvent au niveau de la tête, du cou, des membres, des doigts en particulier, voire des muqueuses.
2- Histologie :
Le plus souvent, c’est dans une veine ectasique que se développent dans la lumière de multiples formations papillomateuses présentant un axe fibreux ou hyalinisé et bordées par une à deux couches de cellules endothéliales.
Les cellules endothéliales sont proéminentes, dépourvues de mitoses et de pléomorphisme.
Quelques foyers de thrombi plus ou moins organisés sont également observés.
Par places, les cellules endothéliales sont présentes dans de larges travées de tissu conjonctif hyalinisé.
Le caractère endothélial des cellules bordantes de la papille est établi par la positivité pour le facteur VIII antigénique.
Quelquefois, le diagnostic différentiel peut se poser avec un angiosarcome, mais la topographie intravasculaire prédominante de la prolifération, l’absence de nécrose, de pléomorphisme net et d’un grand nombre de mitoses plaident plus pour une hyperplasie endothéliale papillaire intravasculaire.
3- Traitement :
Il est chirurgical ou dicté par celui de la lésion sous-jacente.
C – ANGIOME SÉNILE :
Les angiomes séniles touchent, comme leur appellation l’indique, les sujets adultes d’âge moyen et les personnes âgées.
Très fréquentes, ces lésions siègent sur le tronc et tendent à augmenter en nombre et en taille.
Il s’agit de petites élevures asymptomatiques apparaissant parfois sous l’influence de facteurs hormonaux comme la grossesse pour disparaitre dans le post-partum.
Sur le plan histologique, les angiomes séniles correspondent à des capillaires dilatés, avec une paroi vasculaire plus ou moins épaissie.
D – ANGIOME SERPIGINEUX :
L’angiome serpigineux a été décrit à l’origine par Hutchinson, en 1889.
Il est le résultat d’une prolifération de cellules endothéliales aboutissant ainsi à la formation de néocapillaires et non d’une simple dilatation de capillaires préexistants.
1- Clinique :
Il se caractérise par la présence de macules multiples, de petite taille, érythémateuses ou pourpres, évoluant sur plusieurs mois ou années, de façon serpigineuse, parfois regroupées sur un fond érythémateux.
Débutant dans l’enfance, cette lésion asymptomatique est le plus souvent unilatérale et localisée au niveau des membres inférieurs.
Après une période de croissance plus ou moins longue, la lésion se stabilise pendant la vie adulte et peut même régresser complètement, ou du moins en partie.
2- Histologie :
La lésion consiste en un regroupement dans le derme papillaire de capillaires dilatés délimités par des parois épaisses.
E – HÉMANGIOME ACRAL ARTÉRIOVEINEUX :
L’hémangiome acral artérioveineux est un hamartome artérioveineux développé aux dépens du plexus vasculaire souspapillaire et constitué de plusieurs shunts artérioveineux.
Cette lésion touche le sujet adulte (quatrième et cinquième décennies) et se traduit par l’apparition d’une papule bleutée ou rouge, de petite taille, de l’ordre du centimètre, non douloureuse, siégeant à la face, aux muqueuses buccales et aux extrémités.
Sur le plan histologique, elle se caractérise par la présence de multiples vaiseaux dont la majorité, après coloration par l’orcéine, ne présente pas de limitante élastique interne, ou du moins une limitante très altérée.
Les coupes sériées permettent d’objectiver les shunts artérioveineux.
En revanche, on ne peut identifier ni cellules glomiques, ni structures nerveuses infirmant l’hypothèse d’un hamartome du canal de Sucquet-Hoyer.
F – HÉMANGIOME EN CIBLE HÉMOSIDÉRINIQUE :
L’hémangiome en cible hémosidérinique est une entité décrite en 1988 par Santa Cruz et Aronberg.
1- Clinique :
L’aspect clinique très particulier permet une reconnaissance facile de cette lésion papulonodulaire de petite taille inférieure à 1 cm, de couleur pourpre ou violine, entourée d’un double halo clair et ecchymotique lui donnant ainsi un aspect en « cocarde ».
De topographie ubiquitaire (tronc, membres), l’hémangiome en « cible » se présente comme une lésion unique.
Au cours de son évolution, la zone ecchymotique a tendance à régresser et à disparaître, ne laissant que la papule centrale, plus fibreuse et simulant alors un DF.
Ainsi, si l’aspect en « cible » est très évocateur, il n’est ni spécifique, ni obligatoire et présente un caractère transitoire.
Cette tumeur peut donner le change cliniquement avec un nævus mélanocytique, un DF, voire un hémangiome.
2- Histologie :
Les lésions ont un aspect histologique variable en fonction de l’âge de la lésion.
Aux stades précoces, la prolifération mal délimitée est constituée dans le derme papillaire de larges cavités vasculaires, très ectasiques, délimitées par une seule couche de cellules endothéliales, faisant parfois protrusion dans la lumière en « clou de tapissier ».
Par places, ces espaces sont le siège de projections papillaires, amas polypoïdes de cellules endothéliales, oblitérant la lumière du vaisseau.
Des globules rouges, des thrombi de fibrine remplissent également les lumières vasculaires.
Plus en profondeur, dans le derme réticulaire, les espaces vasculaires sont plus irréguliers, anguleux.
Les lumières des vaisseaux sont plus étroites, réduites à l’état de fentes, optiquement vides.
Le collagène apparaît « disséqué » par les structures vasculaires.
Les cellules endothéliales sont alors plus aplaties.
L’extension du processus peut se faire jusqu’au tissu sous-cutané.
Le derme est le siège d’un infiltrat inflammatoire, de globules rouges extravasés.
Les dépôts d’hémosidérine sont fréquents dans ce type de lésion.
Aux stades tardifs, les lumières vasculaires sont collabées, les dépôts d’hémosidérine sont très abondants.
Les espaces extravasculaires sont beaucoup plus cellularisés avec des cellules d’aspect fibroblastique étroitement associées à la prolifération endothéliale.
L’histologie de ces lésions doit faire discuter en premier lieu un sarcome de Kaposi, mais aussi un lymphangioendothéliome bénin.
Les vaisseaux en immunomarquage présentent les marqueurs classiques des cellules endothéliales comme le facteur VIII antigénique et la lectine Ulex europeus.
3- Pathogénie :
La pathogénie de cette lésion n’est pas clairement élucidée.
Cependant, l’hypothèse du traumatisme d’un hémangiome préexistant associé à des phénomènes de thrombose et de recanalisation n’est pas exclue.
Par certains aspects (thrombi de fibrine, projections papillaires), cette tumeur rappelle la tumeur de Masson.
4- Traitement :
Il est chirurgical.
G – HÉMANGIOME MICROVEINULAIRE :
L’hémangiome microveinulaire est une tumeur bénigne vasculaire décrite pour la première fois en 1991 bien que, dès 1989, sous l’appellation d’hémangiome microcapillaire, une tumeur d’aspect histologique identique ait été rapportée.
1- Clinique :
L’hémangiome microveinulaire se présente sous la forme d’une lésion papuleuse ou nodulaire, voire de plaque, de couleur rouge ou pourpre, d’apparition récente (< 3 mois) ou d’évolution lentement progressive sur plusieurs mois, bien délimitée, mesurant entre 0,5 et 2 cm et asymptomatique.
Elle apparaît chez l’adulte jeune ou d’âge moyen.
Elle est le plus souvent solitaire et plus exceptionnellement double.
Cette tumeur touche aussi bien les hommes que les femmes.
La topographie des lésions est ubiquitaire (membres surtout supérieurs, tronc et face). Dans certains cas, des facteurs hormonaux comme la prise de contraceptifs oraux ou la grossesse semblent impliqués dans sa survenue.
Requena et al signalent la possibilité d’observer des hémangiomes de type microveinulaire au cours du syndrome POEMS (Polyneuropathie, Organomégalie, Endocrinopathie, Protéine monoclonale, Anomalies cutanées) (Skinchanges).
2- Histologie :
La tumeur, mal circonscrite, siège dans le derme réticulaire et est constituée de nombreux vaisseaux, arrondis ou ovalaires, anastomosés entre eux de façon irrégulière, remplis pour certains de globules rouges.
Les parois vasculaires sont formées par une ou deux couches de cellules endothéliales, aplaties, parfois légèrement renflées, mais dépourvues de toute atypie.
Il n’existe pas de prolifération endoluminale.
La lumière vasculaire est étroite et parfois collabée.
Les vaisseaux ressemblent à des capillaires ou à de petites veinules.
La prolifération vasculaire se situe au sein d’une trame conjonctive dense faite de faisceaux de collagène épais.
L’infiltrat inflammatoire lymphohistiocytaire est peu marqué. Dans quelques cas, existent de ténus dépôts d’hémosidérine.
Les immunomarquages montrent un marquage classique des cellules endothéliales pour le facteur VIII antigénique et la lectine Ulex europeus.
On retrouve également un marquage positif pour quelques cellules de la paroi vasculaire avec l’actine musculaire lisse.
Cette tumeur est à différencier, sur le plan histologique, du sarcome de Kaposi mais aussi des autres tumeurs vasculaires bénignes acquises, et en particulier de l’angiome en touffes.
H – ANGIOME ACQUIS EN « TOUFFES » :
Wilson Jones est le premier à avoir introduit la dénomination d’angiome en touffes pour désigner une tumeur ayant des particularités histologiques bien définies.
Elle est également connue sous les appellations d’hémangiome capillaire progressif ou d’angioblastome.
1- Clinique :
L’angiome en touffes est une tumeur de l’enfant et de l’adulte jeune, bien que des cas congénitaux ou d’apparition tardive (quatrièmehuitième décennies) aient été rapportés.
Les formes congénitales ou d’apparition très précoce, dans les premiers mois de vie, peuvent être de diagnostic difficile avec un hémangiome.
La lésion se caractérise par sa variabilité dans son expression clinique ainsi que dans sa taille.
Le plus souvent, il s’agit de lésions maculeuses, aux contours mal définis, de couleur rouge terne, brunâtre, avec un aspect parfois en « mottes ».
La surface de la lésion est lisse ou le siège de multiples formations angiomateuses rouge vif.
Il peut exister un léger degré d’infiltration, un aspect surélevé du placard.
L’angiome en « touffes » s’accompagne parfois d’une sensibilité à la palpation, d’une hyperhidrose localisée, d’une légère augmentation de la température locale.
Sa surface est recouverte, dans certains cas, d’un fin lanugo.
Cette tumeur est surtout localisée au niveau du cou des épaules, et de la partie haute du dos.
Elle atteint souvent une taille très importante de plusieurs centimètres.
Une autre de ses caractéristiques est son mode évolutif.
L’extension de la lésion se fait de façon lentement progressive sur plusieurs mois ou années.
Après cette phase d’extension, elle tend à se stabiliser.
Il n’existe pas de tendance à la régression spontanée.
L’angiome en touffes n’a pas de retentissement fonctionnel, si ce n’est dans les exceptionnelles formes s’étendant aux structures profondes.
Le recours à une exploration par scanner ou résonance magnétique nucléaire pour évaluer l’extension en profondeur est indispensable.
L’apparition d’une thrombopénie doit faire évoquer le diagnostic de syndrome de Kasabach-Merritt, d’autant que les travaux d’Enjolras ont bien montré que l’histologie du Kasabach-Merritt peut être celle soit d’un angiome en touffes, soit d’un hémangioendothéliome kaposiforme.
Quelques associations ont été décrites (grossesse, transplantation hépatique…).
2- Histologie :
Le diagnostic d’angiome en touffes n’étant pas aisé sur la simple clinique, le recours à l’histologie s’avère indispensable.
La tumeur est localisée dans le derme moyen et inférieur.
La prolifération tumorale est constituée de touffes arrondies ou ovoïdes de cellules endothéliales situées au sein d’un collagène dermique dépourvu d’anomalies.
La distribution en « boulets de canon » est une des caractéristiques de cette tumeur. Les touffes cellulaires peuvent parfois être coalescentes, donnant des travées de tissu angiomateux.
Les cellules endothéliales sont hypertrophiées, arrondies ou ovoïdes, très étroitement serrées entre elles, ne laissant qu’une lumière vasculaire très étroite. Les mitoses sont rares.
Il n’existe pas d’atypies cellulaires.
L’angiocentricité est un des aspects de cette lésion.
En périphérie des touffes, quelques capillaires renfermant des érythrocytes sont visibles, de même que des fentes et des espaces vasculaires dilatés, en demi-lune, délimités par des cellules endothéliales aplaties.
Il n’existe pas d’infiltrat inflammatoire. De façon exceptionnelle, la tumeur s’étend en profondeur jusqu’au fascia et au muscle.
L’histologie doit aider à différencier ce type de tumeur d’avec un sarcome de Kaposi, un angiosarcome de bas grade bien différencié ou, plus exceptionnellement, de l’angioendothéliome papillaire endovasculaire malin de Dabska où un certain degré d’organisation en touffes est retrouvé mais aussi un aspect lymphocytoïde.
L’immunohistochimie permet d’obtenir un marquage faible ou négatif pour le facteur VIII antigénique et plus marqué pour la lectine Ulex europeus.
3- Traitement :
La chirurgie est parfois rendue difficile par la taille de la lésion.
La radiothérapie, la cryothérapie, l’électrocoagulation ne sont pas des thérapeutiques à conseiller.
Ont été proposées les injections intralésionnelles de corticoïdes ou d’interféron, le laser à colorant pulsé, le recours à des thérapeutiques systémiques comme la corticothérapie générale à fortes doses ou les injections souscutanées d’interféron-alpha. Les résultats restent cependant très aléatoires.







")



{kind=link}