



















Définition – historique :
La STB est une affection caractérisée par la survenue de tumeurs bénignes (hamartomes) dans divers organes.
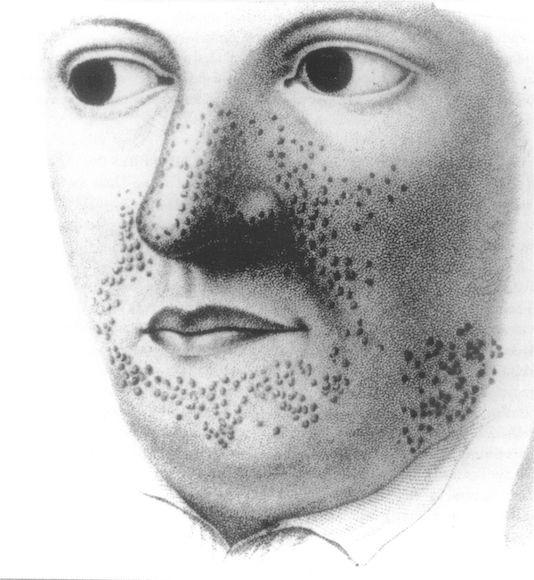 En effet, l’atteinte est pluritissulaire, affectant préférentiellement la peau, le système nerveux central, les reins, le coeur et les poumons.
En effet, l’atteinte est pluritissulaire, affectant préférentiellement la peau, le système nerveux central, les reins, le coeur et les poumons.
La triade classique associant épilepsie, retard mental et « adénomes sébacés » correspond à une forme grave de l’affection ; en effet, l’expressivité de la maladie est très variable et, dans certains cas, le diagnostic est difficile devant la pauvreté des manifestations cliniques.
L’incidence à la naissance est évaluée à 1/5 800, touchant de manière identique les deux sexes.
La transmission de l’affection obéit à un mode autosomique dominant, mais 50 à 70 % des cas sont sporadiques, correspondant à un mécanisme de mutation.
Von Recklinghausen est le premier à avoir rapporté un cas de STB en 1863, mais c’est Bourneville qui a établi un lien entre l’atteinte cutanée et neurologique et a parlé, pour la première fois, de sclérose tubéreuse en 1880.
Les lésions ont alors été décrites en détail par Balzer et Ménétrier en 1885 et Pringle en 1890.
Vogt, en 1908, a complètement intégré la relation neurocutanée et a décrit la triade clinique : épilepsie, retard mental et « adénomes sébacés ».
Manifestations cliniques :
A – MANIFESTATIONS CUTANÉES :
1- Taches hypomélaniques :
Leur fréquence est d’environ 80 à 90 % dans la STB mais elles sont courantes dans la population générale, ce qui leur confère un faible poids diagnostique (0,8 % des nouveau-nés selon Alper ; 4,7 % dans une population adulte dans une étude de Sheryll).
L’âge d’apparition est souvent précoce : les taches existent généralement à la naissance ou, souvent, surviennent lors des premières années de la vie.
Elle sont parfois évidentes cliniquement mais d’autres fois seulement visibles après un examen minutieux en lampe de Wood.
Elles peuvent être uniques ou multiples, localisées à la racine des membres ou sur le tronc, épargnant la face.
Leur taille varie de quelques millimètres à 10 cm.
Elles sont uniformément blanches, sans halo hyperhémique ni hyperpigmentation périphérique.
La sudation n’y est pas abolie et elles rougissent sous frottement, ce qui les distingue des nævi achromiques.
Leur forme est variable : ovalaire, linéaire ou typiquement en « feuille de sorbier ».
Un autre aspect classique est celui d’une hypopigmentation en « confetti », faite de multiples taches de 1 à 2mmde diamètre, distribuées symétriquement sur les membres.
Les taches peuvent se pigmenter légèrement avec l’âge, mais en général elles ne se modifient ni en taille ni en forme.
Histologiquement, le nombre de mélanocytes est normal, avec des mélanosomes de petite taille et en nombre restreint.
2- Angiofibromes :
Ils sont improprement nommés « adénomes sébacés » et sont pathognomoniques de la STB.
Ils se présentent comme des élevures globuleuses de surface lisse, de 1 à 4mmde diamètre, groupées par dizaines ou par centaines, d’une façon symétrique, dans les sillons nasogéniens, qu’ils débordent, sur la racine du nez, dans le sillon mentonnier et sur la zone médiane du menton.
Plusieurs cas d’angiofibromes unilatéraux ont été décrits, le mécanisme génétique pourrait être une mutation postzygotique.
Certaines lésions sont de la couleur de la peau normale ou jaunâtres, mais la plupart sont rouges et couvertes de fines télangiectasies.
On distingue classiquement deux types :
– le type Pringle, de couleur rose ou rouge et de consistance élastique, à prédominance vasculaire ;
– le type Hallopeau-Leredde, variété dure à prédominance fibreuse.
Les angiofibromes sont rarement présents à la naissance, ils apparaissent habituellement entre 5 et 10 ans, ou même plus tardivement.
Ils persistent ensuite indéfiniment, avec des poussées à la puberté et pendant les grossesses, sans jamais dégénérer.
3- Plaques « peau de chagrin » :
Elles apparaissent généralement entre 2 et 5 ans, parfois plus tardivement.
En général unique, siégeant préférentiellement dans la région lombosacrée, la lésion se présente comme une plaque discrètement surélevée de surface fripée, granitée ou d’aspect pavé, de couleur jaunâtre ou identique à celle de la peau normale.
Sa taille varie de quelques millimètres à une dizaine de centimètres.
4- Fibromes périunguéaux de Koënen :
Ils sont caractéristiques de l’affection et parfois en sont les seules manifestations.
Ils doivent donc être recherchés très minutieusement.
Ce sont des petites proliférations charnues, grises ou rosées, fermes, se détachant en bas du sillon périunguéal et poussant parallèlement à l’ongle, qu’elles dépriment parfois en gouttière longitudinale.
Elles sont oblongues, en « grain de blé », leur extrémité distale est souvent cornée.
Les fibromes siègent plus souvent au niveau des orteils que des doigts, généralement uniques par ongle mais parfois au nombre de deux ou trois.
L’évolution peut se faire vers la nécrose spontanée ou traumatique, mais la tumeur ne dégénère jamais.
5- Autres manifestations cutanées :
D’autres lésions sont retrouvées chez les patients atteints de STB :
– des fibromes de la racine des cheveux ou des sourcils ;
– des molluscum pendulum ;
– des plaques fibreuses frontales qui peuvent être la première manifestation de la maladie. Les taches « café au lait » ne sont pas plus fréquentes que dans la population générale.
L’atteinte des muqueuses est également possible ; en effet, les muqueuses buccales (gencive antérieure surtout, voile du palais, langue) sont assez souvent le siège de petits éléments miliaires en semis, de nodules fibreux ou d’infiltrations diffuses.
Les phanères peuvent également être atteintes, cela se manifeste par :
– des mèches de cheveux gris (poliose) ou blanches (leucotrichie).
Celles-ci peuvent être la manifestation la plus précoce de la maladie, en étant visibles dès la naissance ;
– des défauts ou « trous » de l’émail dentaire.
Ceux-ci peuvent se voir sur les dents de lait tout comme sur les dents définitives (incisives et canines en particulier) ; ils peuvent être un argument diagnostique complémentaire lors de la recherche d’une sclérose tubéreuse.
B – MANIFESTATIONS NEUROLOGIQUES :
L’atteinte du système nerveux central est responsable de la majorité de la morbidité et de la mortalité dans la STB.
L’épilepsie et le retard mental en sont les principales manifestations.
Les lésions cérébrales sont de trois types :
– les tubérosités corticales ou sous-corticales ;
– les nodules sous-épendymaires ;
– les astrocytomes à cellules géantes.
Ces lésions sont multifocales, distribuées dans les hémisphères cérébraux mais aussi dans le cervelet et le tronc cérébral.
Il est très difficile d’évaluer la prévalence des différentes manifestations neurologiques en raison de l’important biais de recrutement.
1- Épilepsie :
C’est la manifestation la plus fréquente de la STB avec une prévalence de 62 à 92%.
L’épilepsie peut survenir de manière isolée mais est plus fréquemment associée à un retard mental.
Elle peut survenir à tout âge, de la période néonatale à l’âge adulte.
Le type le plus précoce et le plus caractéristique d’épilepsie en cas de STB est le syndrome de West (spasmes infantiles).
Il est fréquemment le mode de révélation de la STB (70 % des cas), mais la STB n’est responsable que de 5 à 7% des syndromes de West.
Le diagnostic repose sur la constatation de la triade de West qui associe :
– des clonies axiales ou spasmes, le plus souvent en flexion survenant par brefs accès ;
– une régression psychomotrice ;
– un tracé d’hypsarythmie à l’électroencéphalogramme (EEG).
Celui-ci survient en général entre 3 et 10 mois. Son pronostic est généralement sombre et la réponse au traitement souvent incomplète.
Avec la maturation corticale et sous-corticale, les spasmes se résolvent parfois spontanément mais sont souvent alors remplacés par d’autres types de crises convulsives, partielles ou généralisées.
Dans 55 à 60 % des cas, l’évolution se fait vers un syndrome de Lennox-Gastaut diagnostiqué sur les critères suivants :
– début entre 1 et 7 ans ;
– multiples types d’épilepsie ;
– prévalence importante des difficultés d’apprentissage (plus de 90 %) ;
– EEG : longues salves de pointes-ondes à deux cycles/seconde.
La STB est la cause de 2 % des Lennox-Gastaut.
L’histoire naturelle de l’épilepsie dans la STB après le syndrome de West et le Lennox-Gastaud se poursuit fréquemment par une augmentation, en fréquence et en gravité, des crises convulsives dans la petite enfance, avec un échappement au traitement.
On remarque souvent, en revanche, à l’adolescence et au début de l’âge adulte, une diminution spontanée de la fréquence des crises.
Plus rarement, l’épilepsie est due à des tumeurs cérébrales ; c’est l’hypothèse qu’il faut envisager chez un sujet qui fait, pour la première fois, une crise partielle, ou chez qui la fréquence des crises se majore.
2- Retard mental et difficultés d’apprentissage :
Les capacités intellectuelles des patients atteints de STB sont très variables, allant du développement rigoureusement normal jusqu’à l’arriération mentale profonde.
La prévalence du retard d’apprentissage varie de 38 % à 80%.
S’il ne semble pas exister de rapport avec l’atteinte cutanée, on note, en revanche, une relation directe entre de sévères difficultés d’apprentissage et l’épilepsie, tout particulièrement si :
– les crises convulsives surviennent précocement (spasmes infantiles en général) ;
– elles persistent et évoluent vers un syndrome de Lennox-Gastaut ;
– elles sont difficiles à contrôler par le traitement.
Le dysfonctionnement intellectuel se manifeste en général assez précocement, dès les premiers mois de vie.
Beaucoup plus rarement, l’évolution semble normale initialement, les difficultés survenant à l’adolescence ou à l’âge adulte.
3- Troubles du comportement :
Ils sont fréquents dans la STB, en particulier l’autisme existant chez 50 % des sujets atteints de STB.
Les autres manifestations classiques sont l’hyperactivité, l’agressivité et les automutilations.
Ces troubles sont d’origine multifactorielle :
– directement liés au dysfonctionnement cérébral ;
– conséquences du retard d’apprentissage ;
– liés à l’usage simultané de plusieurs antiépileptiques ; – ou, plus rarement, séquelles d’une épilepsie mal contrôlée.
4- Troubles du sommeil :
On les retrouve chez 60 % des patients atteints de STB ; ils se manifestent par des retards à l’endormissement ou des réveils fréquents.
La pathogénie est difficile à déterminer, tout comme ses rapports avec l’épilepsie, le retard d’apprentissage ou les troubles du comportement.
Cependant, il est clair que des troubles du sommeil peuvent engendrer des difficultés d’apprentissage et une majoration de troubles du comportement.
5- Tumeurs cérébrales et hydrocéphalie :
Les nodules sous-épendymaires ou les tubérosités peuvent entraîner une obstruction du trou de Monro ou, moins fréquemment, de l’aqueduc, responsable alors d’une hydrocéphalie.
Plus rarement, les tubérosités peuvent se transformer en astrocytomes à cellules géantes ou glioblastomes (5 à 10 % des patients).
Cette transformation se traduit par un syndrome d’hypertension intracrânienne, ou par la survenue de crises partielles.
6- Imagerie cérébrale :
Actuellement, l’examen le plus performant pour mettre en évidence les lésions cérébrales de STB est l’imagerie par résonance magnétique (IRM). Le scanner n’a de supériorité que pour les lésions calcifiées.
Les lésions visibles sont de quatre types :
– tubérosités corticales et sous-corticales : présentes dans 95 % des cas.
En IRM, il existe un iso- ou hyposignal en T1 et un hypersignal en T2 (sauf chez le nouveau-né où les images sont inversées).
L’injection de gadopentetate dimeglumine peut accentuer les signaux.
Au scanner, les lésions sont hypodenses, le seul intérêt de cet examen est de montrer des calcifications non visibles à l’IRM ;
– lésions de la substance blanche : sous forme de bandes linéaires s’étendant de l’épendyme aux tubérosités, elles sont retrouvées chez 90 % des patients et leur nombre est proportionnel à celui des tubérosités ;
– nodules sous-épendymaires : présents chez 95 % des patients atteints de STB et généralement bilatéraux.
Leur évolution se fait progressivement vers la calcification, ce qui explique la supériorité du scanner sur l’IRM pour les mettre en évidence.
Ils sont responsables d’un iso- ou hyposignal en T1 et d’un hyposignal en T2 à l’IRM.
La prise de contraste après injection est classique et ne signe pas forcément une transformation néoplasique ;
– astrocytomes à cellules géantes : survenant chez environ 15 % des patients atteints de STB et en général localisés à proximité du trou de Monro.
L’aspect des lésions en IRM est superposable à celui des nodules sous-épendymaires.
Il est donc conseillé de réaliser des IRM de manière annuelle chez les patients ayant des images suspectes, et en particulier entre 8 et 18 ans, âges où la survenue d’astrocytome est la plus fréquente.
Une détection précoce facilitera, en cas d’indication, l’acte chirurgical.
L’évaluation de l’importance des lésions cérébrales présente, par ailleurs, l’intérêt de permettre une extrapolation clinique.
En effet, quand le nombre de tubérosités est élevé, le retard mental, les spasmes infantiles et la survenue précoce de l’épilepsie sont plus fréquents.
C – MANIFESTATIONS OPHTALMOLOGIQUES :
L’atteinte ophtalmologique la plus fréquente dans la STB est le phacome rétinien, il s’observe dans environ 50 % des cas.
Histologiquement, il s’agit d’un hamartome astrocytaire qui peut prendre trois formes cliniques :
– la forme plane qui se présente comme un placard grisâtre, gélatineux, lisse, aux bords flous ;
– la forme saillante, muriforme, qui correspond à une tumeur blanc grisâtre, en amas de « grains de tapioca » ;
– la forme intermédiaire. Le diagnostic de ces tumeurs est ophtalmoscopique.
Les lésions étant en général asymptomatiques, le diagnostic est fait de façon fortuite ou lors d’un examen systématique chez un sujet ayant d’autres signes de la maladie. L’angiographie fluorescéinique met en évidence la composante vasculaire des lésions.
En effet, on note un remplissage rapide des vaisseaux au temps artériel, il persiste ensuite une hyperfluorescence aux temps tardifs.
Les lésions n’entraînent en général pas de signes fonctionnels, la baisse d’acuité visuelle est rare, elle peut être due à une hémorragie du vitré qui fait partie des rares complications possibles. Les phacomes rétiniens sont classiquement peu évolutifs et ils n’ont pas de traitement spécifique.
D’autres manifestations ophtalmologiques peuvent être observées : des zones de dépigmentation rétinienne ou irienne, des nodules fibreux sous-conjonctivaux ou palpébraux.
D – MANIFESTATIONS RÉNALES :
Dans la STB, la principale manifestation rénale est l’angiomyolipome.
Il s’agit d’une tumeur bénigne faite de muscle lisse, de tissu adipeux et de vaisseaux sanguins. Sa taille varie de 1 à 15 cm.
Elle siège préférentiellement au niveau cortical.
C’est une tumeur rare, qui représente environ 3 % des tumeurs solides du rein.
Son histoire naturelle permet de distinguer deux formes :
– la forme sporadique : 80 % des cas, où les tumeurs sont en général peu nombreuses, de petite taille et asymptomatiques ;
– la forme associée à la STB (20 %), où les tumeurs sont multiples et bilatérales.
Des angiolipomes existent chez 40 à 80 % des patients atteints de STB.
Les lésions peuvent rester asymptomatiques et être découvertes fortuitement lors d’une échographie abdominale.
Dans d’autres cas, elles sont révélées par des douleurs abdominales ou du flanc, une masse palpable, une hématurie ou une anémie.
Le principal risque évolutif des angiomyolipomes est une hémorragie (5 à 25 % des cas). Ce risque est d’autant plus important que l’angiomyolipome est de grande taille (> 4 cm) et symptomatique.
La grossesse est une période particulièrement à risque de saignement.
Dans environ 16 % des cas, les angiomyolipomes peuvent se compliquer d’hypertension artérielle (HTA).
L’évolution peut également se faire vers l’insuffisance rénale (13 % des cas).
Même si l’angiomyolipome est une tumeur évolutive qui augmente de taille (67 % des cas selon Steiner), il n’est jamais constaté de dégénérescence maligne. L’échographie rénale est l’examen clef pour le diagnostic d’angiomyolipome.
Celui-ci se présente comme une tumeur homogène, hyperéchogène (en raison de la composante graisseuse).
Cette image n’est pas spécifique, en effet certains adénocarcinomes rénaux peuvent prendre le même aspect.
Il est donc nécessaire de confirmer le diagnostic d’angiomyolipome rénal par un scanner en coupes fines sans, puis avec injection de produit de contraste.
La tumeur est hypodense, avant et après l’injection.
L’IRM est réservée aux cas douteux.
L’artériographie rénale n’est indiquée qu’avant l’embolisation d’une artère principale ou avant un geste chirurgical sur un angiomyolipome de grande taille.
Le traitement de l’angiomyolipome doit être le plus conservateur possible.
La taille et le caractère évolutif sont les deux principaux éléments qui guident l’approche thérapeutique.
En effet, les tumeurs dont la taille est inférieure à 3,5 ou 4 cm, ou asymptomatiques, peuvent être simplement surveillées par échographie tous les 6 mois.
En revanche, si la taille de la tumeur excède 4 cm, ou si elle est symptomatique ou évolutive, une chirurgie doit être envisagée (la plus conservatrice possible), l’embolisation peut être aussi une bonne solution.
Chez les patients atteints de STB, l’angiomyolipome peut être associé à d’autres lésions rénales :
– carcinome à cellules rénales, dont l’incidence est diversement appréciée (26 % selon Tanaka et al, nettement moindre dans une étude récente de Steiner) ;
– des kystes rénaux multiples ;
– une polykystose rénale (4 %), l’évolution vers l’insuffisance rénale est alors classique.
E – MANIFESTATIONS CARDIAQUES :
La STB peut s’accompagner de rhabdomyomes cardiaques qui s’expriment plus particulièrement chez le nouveau-né.
Les tumeurs cardiaques primitives sont rares dans la population générale et très inhabituelles chez l’enfant.
Parmi celles-ci, la plus fréquente chez le nouveau-né (deux tiers des cas) est le rhabdomyome. Il est classiquement reconnu que les rhabdomyomes cardiaques sont associés à une STB dans 50 % des cas.
Inversement, chez environ 50 % des patients atteints de STB, on retrouve des rhabdomyomes, mais ce chiffre est très variable au cours de la vie ; en effet, les rhabdomyomes apparaissent très précocement en période anténatale et ont une tendance spontanée à la régression.
L’incidence des rhambdomyomes à la naissance chez les patients atteint de STB serait de 58 % et diminuerait rapidement pour atteindre 18 % à l’âge adulte.
Il s’agit souvent de la manifestation la plus précoce de la STB.
Le diagnostic peut être fait en période anténatale par échographie de routine, mais l’arythmie foetale est le mode de révélation le plus habituel, conduisant à la réalisation d’une échographie foetale qui fait le diagnostic.
Dans d’autres cas, la détection prénatale est faite chez des foetus à risque de STB.
Le diagnostic de rhabdomyome est fait au mieux échographiquement ; en effet, il existe une bonne corrélation entre les images échographiques et l’histologie, les autres tumeurs cardiaques primitives (tératomes, myomes et fibromes) étant très différentes échographiquement.
Le rhabdomyome se présente macroscopiquement sous la forme de nodules blanc-gris souvent multiples, disséminés en règle dans les deux ventricules, voire infiltrant de façon diffuse le myocarde.
Si les rhabdomyomes sont souvent asymptomatiques, ils peuvent aussi être responsables d’obstruction ou de fuite valvulaire entraînant une insuffisance cardiaque ou des troubles du rythme tels que le syndrome de préexcitation ventriculaire, Wolff-Parkinson-White, flutter auriculaire (FA), bloc atrioventriculaire, extrasystoles et tachycardie ventriculaire.
La majorité des rhabdomyomes régressent spontanément, ce qui est un plaidoyer en matière de traitement pour une abstention ou un simple traitement médical des troubles du rythme.
Seuls les rhambdomyomes responsables d’obstruction, de troubles hémodynamiques ou d’arythmie résistant au traitement devront bénéficier de la chirurgie.
F – MANIFESTATIONS PULMONAIRES :
L’atteinte pulmonaire dans la STB est rare (1 % des cas selon Dwyer), de révélation souvent tardive, elle touche essentiellement les sujets de sexe féminin.
Histologiquement, l’atteinte est identique à celle de la lymphangiomyomatose ; elle se caractérise par l’existence de nombreux kystes parenchymateux dont les parois sont constituées de muscle lisse.
Selon la localisation de cette prolifération musculaire, les signes cliniques varient :
– autour des voies respiratoires, elle entraîne une obstruction, la formation de bulles et de pneumothorax ;
– autour des veines et veinules : une dilatation responsable d’hémoptysie ;
– autour des artères : une HTA pulmonaire et un coeur pulmonaire ;
– autour des lymphatiques : une obstruction et un chylothorax.
La forme pulmonaire touche avec prédilection les femmes (84 ou 74 % selon Dwyer) en période d’activité génitale (âge moyen de diagnostic des lésions pulmonaires : 34 ans).
Les symptômes cliniques sont en général d’apparition progressive.
Le signe d’appel le plus fréquent est la dyspnée d’effort (68 %) pouvant s’associer à une toux (27 %) ou à des hémoptysies (27 %).
L’évolution se fait ensuite lentement vers l’insuffisance respiratoire, avec coeur pulmonaire chronique. L’évolution est également fréquemment émaillée par la survenue de pneumothorax volontiers récidivants et bilatéraux.
Différents examens complémentaires peuvent aider au diagnostic de l’atteinte pulmonaire ou de sa sévérité :
– la radiographie pulmonaire objective classiquement un syndrome interstitiel diffus avec un aspect réticulé ou réticulonodulaire.
Des images en « rayon de miel » ou des kystes peuvent également être visibles en cours d’évolution.
La radio pulmonaire permet également de faire le diagnostic de pneumothorax ;
– le scanner thoracique permet de dépister les lésions pulmonaires à un stade plus précoce et même asymptomatique.
Il ne permet pas, en revanche, de différencier ces lésions de celles de la lymphangioléiomyomatose ;
– les épreuves fonctionnelles respiratoires sont très utiles pour évaluer la gravité de l’atteinte pulmonaire ; il existe en général un syndrome obstructif.
Les gaz du sang artériel peuvent montrer une hypoxie de repos.
Le transfert du CO peut être réduit.
Les liens entre l’atteinte pulmonaire dans la STB et la lymphangioléiomyomatose sont très controversés : pour Stovin et al en 1975, il s’agissait de deux affections différentes ; pour Maziak et al, en 1996, il s’agit de deux aspects différents d’une même maladie. ; Castro et al, en 1995, concluent que la STB et la lymphangiomyomatose sont semblables en termes d’histoire naturelle, de manifestations cliniques et de réponse au traitement.
Tout récemment, Smolarek et al ont mis en évidence, chez sept femmes atteintes de lymphangiomyomatose isolée, une perte d’hétérozygotie de TSC2.
Il est indéniable que ce sont deux entités très proches dont les liens ne sont pas encore définis précisément.
Le traitement des manifestations pulmonaires de STB était, jusqu’ici, essentiellement symptomatique.
Les importantes similitudes avec la lymphangiomyomatose ont encouragé à essayer des traitements hormonaux, tout comme dans la lymphangiomyomatose.
La progestérone et le tamoxifène ont donné des résultats variables.
Le traitement de choix actuel est l’injection mensuelle de médroxyprogestérone.
L’ovariectomie et le tamoxifène peuvent être des alternatives thérapeutiques.
G – AUTRES MANIFESTATIONS :
L’atteinte hépatique est moins bien connue car presque toujours asymptomatique, mais la fréquence des hamartomes hépatiques est loin d’être négligeable (45 % des sujets de plus de 10 ans selon Joswiak et al). Histologiquement, il s’agit d’un angiomyolipome, tumeur mésenchymateuse à triple composante : vasculaire, adipeuse et musculaire lisse.
Cette tumeur est rare en dehors de la STB.
Elle est en général asymptomatique et sans répercussion biologique.
En échographie, l’image classique est celle d’une lésion bien limitée, hyperéchogène et hétérogène ; en tomodensitométrie (TDM), les lésions sont hypodenses et hétérogènes.
La preuve diagnostique est apportée par la cytoponction.
L’abstention thérapeutique est la règle.
Des microhamartomes rectaux surviennent chez deux tiers des patients atteints de STB, ils sont en général multiples et asymptomatiques.
Des désordres endocriniens ont également été décrits : puberté précoce, diabète insipide, déficit en growth hormone (GH).
Les localisations osseuses de la STB sont en général asymptomatiques.
Elles se manifestent radiologiquement par des zones ostéocondensées au niveau de la voûte crânienne, du rachis, des côtes ou du bassin, correspondant à un tissu fibreux.
Critères diagnostiques et pronostiques :
L’expression clinique de la STB est caractérisée par son polymorphisme et sa variabilité d’un sujet à l’autre.
Le diagnostic peut être évident dans les formes classiques : épilepsie, retard mental, signes cutanés, mais, bien souvent, l’expression est plus discrète et il est alors important de reconnaître, à l’aide de critères cliniques stricts, les signes de la maladie pouvant affirmer la sclérose tubéreuse.
Ce diagnostic engage le médecin à plusieurs niveaux :
– diagnostique : pour une prise en charge précoce de la maladie ;
– pronostique : par la recherche de localisations encore asymptomatiques ;
– préventif : au niveau familial par le conseil génétique ;
– pathogénique : dans le cadre d’une corrélation génotype-phénotype.
Ces critères sont cliniques, radiologiques ou histologiques.
Selon leur spécificité dans la STB, leur poids diagnostique varie, ce qui permet de définir des critères primaires, secondaires ou tertiaires.
Il est essentiel de remarquer que toutes ces manifestations ne surviennent pas au même âge et n’ont pas le même profil évolutif.
Ainsi, en période néonatale, le problème cardiaque est au premier plan avec la survenue de troubles du rythme ou la découverte systématique de rhabdomyomes.
Chez l’enfant, l’épilepsie et le retard mental sont un mode classique d’entrée dans la maladie.
Les taches dépigmentées peuvent également être les premières manifestations, si elles ne sont pas spontanément visibles, il convient de les rechercher par examen à la lampe de Wood.
Les angiofibromes de la face se multiplient à la puberté.
À l’âge adulte apparaissent les kystes rénaux et les angiomyolipomes hépatiques et rénaux.
Génétique :
A – INTRODUCTION :
La STB présente une hétérogénéité génétique et clinique.
Deux gènes responsables de STB ont été décrits et, pour une même mutation, on observe un spectre clinique très large.
En 1991, des études de liaisons ont révélé un troisième locus mais cette observation a été infirmée.
B – ORGANISATION ET FONCTION DE TSC1 ET TSC2 :
La physiopathologie de la STB est très mal comprise et de gros efforts dans ce sens ont été entrepris en clonant TSC1 en 9q34 et TSC2 en 16p13.3. Le clonage positionnel de TSC2 (1993) a été facilité par la mise en évidence de larges délétions en 16p13.3.
Celui de TSC1 (1997) fut plus délicat car, en 9q34, on n’observe pas ce type de délétions ; il s’est écoulé 10 ans entre la première localisation et le clonage de TSC1 car un séquençage extensif de la région candidate a dû être réalisé. TSC1 possède un acide ribonucléique messager (ARNm) de 8 600 paires de base (8,6 kb) et code pour une protéine de 130 kDa : l’hamartine. TSC2, composé de 45 exons, occupe 35 kb et code pour la tubérine (130 kDa).
La tubérine comporte un domaine GTP-ase activating proteine (GAP) et activateur transcriptionnel.
À l’inverse, l’hamartine n’a révélé aucune homologie avec d’autres protéines.
Ces deux protéines auraient un rôle dans la même voie signalétique.
Leur dimérisation prédite à partir de leur séquence protéique a été confirmée par des expériences in vivo. TSC2 est un gène d’expression quasi ubiquitaire et on ne note pas de différence quantitative entre l’expression dans des tissus porteurs de la mutation et celle de tissus homozygotes normaux.
C – MÉCANISME TUMORAL :
La STB est une phacomatose et il semble clair maintenant que l’apparition des tumeurs hamartomateuses est liée à une perte d’hétérozygotie (LOH).
C’est-à-dire qu’en plus de la mutation d’un allèle de TSC1 ou TSC2 hérité d’un des deux parents, une seconde mutation (en général une délétion) survient de façon aléatoire sur le second allèle au cours de la vie du patient.
La tumeur, dans ce cas, est due à une anomalie récessive de TSC1 ou TSC2, mais le type de transmission observé est autosomique dominant.
Ces observations tendent à prouver que TSC1 et TSC2 sont des gènes supresseurs de tumeur.
Cette hypothèse a été étayée par plusieurs études effectuées sur des hamartomes (angiomyolipomes, rhabdomyomes, astrocytomes).
Au total, l’équipe de AJ Green a observé une LOH dans 21 tumeurs sur 51 étudiées (41 %) ; 16 impliquaient le locus TSC2 et cinq celui de TSC1.
La corrélation entre la prévalence des angiomyolipomes et l’âge du patient renforce l’hypothèse du gène supresseur de tumeur.
Enfin, les travaux de Kobayachi et al sur TSC2 (transgenèse sur le rat d’Eker) confirment les études citées (cf supra).
Le rat d’Eker est atteint d’un carcinome rénal à transmission autosomique dominante lié à une mutation de TSC2.
Par transgenèse, Kobayashi et al ont montré qu’en rajoutant une troisième copie non mutée de TSC2 au génome du rat d’Ekert, on n’observait pas l’apparition de carcinome.
D – RÉPARTITION DES MUTATIONS ET CORRÉLATION PHÉNOTYPE-GÉNOTYPE :
Selon les études de liaisons, les cas familiaux de STB ségrégeraient avec les deux loci en proportion équivalente.
Toutefois l’étude de AC Jones et al montre une prédominance nette de TSC1 dans les cas familiaux et TSC2 dans les cas sporadiques.
1- TSC1 :
Il est beaucoup plus présente dans les formes familiales (37 % des mutations retrouvées) que dans les formes sporadiques (8 %).
Au niveau de TSC1, il n’y a pas de délétion.
Les mutations ponctuelles, responsables de protéines tronquées dans la plupart des cas, sont également réparties de façon homogène ; très peu sont récurrentes : trois mutations retrouvées chez deux individus non apparentés dans une étude sur 169 cas.
2- TSC2 :
Dans une étude portant sur 90 patients atteints de STB, il a été retrouvé des mutations pathogènes du gène TSC2 dans 32 % des cas sporadiques et 9 % des cas familiaux.
Le long de TSC2, les mutations sont distribuées de façon homogène et tous les types sont représentés : délétions, insertions, duplications, non sens, faux sens.
3- Corrélation phénotype génotype :
AC Jones et al ont remarqué que le retard mental était significativement plus fréquent lorsqu’il s’agissait de néomutations (le plus souvent associée à TSC2).
Mais le biais de recrutement entre les deux populations (familiales et sporadiques) est possible.
En dehors de cette observation, il n’y a pas de corrélation phénotype génotype évidente : ni les mutations responsables d’un décalage du cadre de lecture, ni celles touchant le domaine GAP ou activateur transcriptionnel (pour TSC2) ne sont responsables de formes plus graves.
E – CAS PARTICULIER DE LA POLYKYSTOSE RÉNALE :
Les kystes rénaux surviennent dans approximativement un tiers des patients atteints de STB, mais la polykystose rénale sévère du nourrisson est beaucoup plus rare.
Le gène de la polykystose autosomique dominante PKD1 a été identifié en 1994 et est situé en 16p, directement adjacent à TSC2.
Les deux gènes sont orientés en sens inverse.
Pour la première fois, Brook-Cater et al ont mis en évidence, chez six patients atteints de polykystose rénale sévère d’apparition précoce, une délétion de TSC2 et PKD1.
La même équipe, sur une série plus importante de 27 patients, a montré que cette même délétion n’a jamais été retrouvée chez les patients atteints de kystes survenus à l’âge adulte et aucun patient porteur d’une mutation respectant PKD1 n’a souffert de polykystose sévère précoce, mis à part cinq cas porteurs d’une très large délétion de TSC2 isolée (affectant possiblement l’expression de PKD1).
Il a été aussi mis en évidence des patients mosaïques, avec des atteintes de sévérité variable.
Il s’agit donc d’un syndrome de gènes contigus avec un phénotype original résultant d’une synergie des deux gènes.
F – CONSEIL GÉNÉTIQUE :
Dans la STB, il se heurte à la variabilité d’expression, problème fréquemment rencontré dans les maladies autosomiques dominantes.
Quant à la non-pénétrance, elle est rare, et on ne peut l’affirmer qu’après un examen clinique et paraclinique complet.
En ce qui concerne le conseil génétique dans la STB, il existe deux grands cas de figure :
– la forme familiale connue où le risque de transmission est de un sur deux ;
– la forme sporadique où la difficulté consiste à ne pas manquer le diagnostic de STB chez un des parents atteint d’une forme très discrète.
Cela pose le problème de la non-pénétrance dans la STB. Dans la forme sporadique vraie, le risque de récurrence est de 2 %.
En anténatal, le diagnostic prénatal (DPN) est possible s’il s’agit d’une forme familiale et que la mutation a été identifiée chez les parents.
Toutefois, l’expressivité variable de la STB ne permet pas d’évaluer la sévérité de l’atteinte de l’enfant à naître.
Dans une forme sporadique, il semble licite d’effectuer un DPN (cas d’une mutation identifiée chez un premier enfant de parents sains) pour écarter tout risque de récurrence dans le cas d’une mosaïque germinale parentale.
L’imagerie ne peut affirmer ou infirmer un diagnostic de STB.
Mais, selon Sonigo et al, 87 % des foetus porteurs de rhabdomyomes (visualisés en échographie généralement au troisième trimestre) sont atteints de STB.
L’IRM foetale a développé, en plus du diagnostic, des éléments pronostiques en détectant les nodules subépendymaux.
En présence de ces lésions cérébrales diffuses, le pronostic neurologique serait défavorable.
G – CONCLUSIONS POUR LA PRATIQUE CLINIQUE :
La confirmation par biologie moléculaire d’un diagnostic clinique de STB sera difficile en raison :
– de l’hétérogénéité génétique : deux gènes pour une même maladie ;
– de la taille importante de TSC2 et TSC1 ;
– de la diversité des mutations et la quasi-absence de mutations récurrentes ;
– de l’absence de « points chauds » : la recherche de mutation n’est pas orientée ;
– du fait que plus de la moitié des mutations de TSC2 et la quasitotalité des mutations de TSC1 conservent le cadre de lecture et ne sont pas détectables par la PTT (protein truncating test).
En dehors de la délétion emportant PKD1 et TSC2, la découverte d’une mutation de TSC1 ou TSC2 en anténatal ou chez un nourrisson ne permettra pas de se prononcer sur la gravité du handicap, et notamment sur la présence d’une comitialité ou d’un retard mental.







")



{kind=link}