



















Introduction :
Le sarcome d’Ewing est une tumeur osseuse primitive maligne de l’enfant et l’adolescent, plus rarement de l’adulte.
 Elle est caractérisée par des amas denses de petites cellules rondes en nappes, sans différenciation tissulaire et fait partie de la famille des tumeurs neuroectodermiques qui présentent une translocation 11-22 caractéristique.
Elle est caractérisée par des amas denses de petites cellules rondes en nappes, sans différenciation tissulaire et fait partie de la famille des tumeurs neuroectodermiques qui présentent une translocation 11-22 caractéristique.
Elle peut atteindre tous les os du squelette.
C’est une lésion lytique, avec un envahissement fréquent des parties molles.
En l’absence de traitement, l’évolution fatale est habituellement rapide, avec apparition de métastases à distance, pulmonaires ou osseuses.
Le traitement actuel repose sur l’association d’une chimiothérapie et d’un traitement local chirurgical, parfois complété d’une radiothérapie.
Le pronostic dépend de l’existence de métastases et de la réponse à la chimiothérapie.
La probabilité de survie sans récidive à 5 ans est actuellement de 70 % dans les formes localisées.
Les protocoles les plus récents tentent, grâce à des chimiothérapies à hautes doses, d’améliorer ces résultats dans les formes de mauvais pronostic.
Les progrès de la biologie moléculaire peuvent faire espérer de nouveaux traitements dans l’avenir.
A – DÉFINITION. TERMINOLOGIE :
Le sarcome d’Ewing est une tumeur osseuse primitive maligne, caractérisée par des amas denses de petites cellules rondes uniformes en nappes, sans structure tissulaire reconnaissable.
La première description d’une tumeur osseuse primitive ressemblant au myélome multiple, particulièrement sensible à la radiothérapie, et qu’il avait nommée endothéliome diffus ou myélome endothélial, a été attribuée à Ewing en 1921.
Pour Huvos, des lésions comparables avaient déjà été décrites par Lücke, dès 1866.
L’origine du sarcome d’Ewing a été longtemps source de controverses.
On a parlé de sarcome réticuloendothélial, certains ont pensé qu’il pouvait s’agir de métastases de neuroblastome et on a longtemps considéré qu’il provenait du mésenchyme indifférencié. Turc-Carel et Aurias, qui ont décrit la translocation 11-22, ont permis d’établir que le sarcome d’Ewing était d’origine neuroectodermique, comme les tumeurs neuroectodermiques périphériques (PNET), les neuroépithéliomes et les tumeurs d’Askin de la paroi thoracique.
Ces tumeurs de caractéristiques communes en biologie moléculaire forment le groupe des tumeurs neuroectodermiques ou famille des tumeurs d’Ewing.
B – ÉPIDÉMIOLOGIE. FRÉQUENCE. ÂGE. SEXE :
Le sarcome d’Ewing touche surtout les enfants ou les adolescents et il est rare chez l’adulte après 30 ans.
Le patient le plus jeune rapporté avait 5 mois et le plus âgé avait 83 ans.
Le pic d’incidence se situe entre 5 et 9 ans chez les filles et entre 10 et 14 ans chez les garçons.
Environ 80 % des cas surviennent dans la seconde décennie. Les garçons sont plus souvent atteints que les filles, avec un sex-ratio d’environ 6 pour 4.
Le sarcome d’Ewing est la deuxième tumeur osseuse maligne primitive de l’enfant, après l’ostéosarcome.
Il s’agit néanmoins d’une tumeur assez rare et l’on estime son incidence à environ 50 à 80 cas/an en France.
Il représente 5 à 15% de l’ensemble des tumeurs osseuses malignes primitives.
Il est plus fréquent chez les sujets d’origine européenne, qui représentent environ 96 % des cas et il est exceptionnel chez les sujets d’origine africaine ou afroaméricaine (1,8 %) ou d’autres origines (2,2 %).
C – ÉTIOLOGIE :
L’étiologie précise du sarcome d’Ewing n’est pas encore connue mais il existe probablement des facteurs héréditaires puisqu’il touche préférentiellement les Blancs et que des formes familiales ont été décrites.
Des travaux récents, mais controversés, pourraient indiquer que certaines infections virales joueraient un rôle dans l’induction des transformations génétiques nécessaires au développement des tumeurs d’Ewing.
Les techniques de cytogénétique, d’immunocytochimie et de biologie moléculaire ont permis de confirmer l’origine neurale du groupe des tumeurs d’Ewing.
Actuellement, on pense que le sarcome d’Ewing a pour origine les cellules parasympathiques postganglionnaires, expliquant les nombreuses localisations possibles de ces tumeurs.
Différents caractères le distinguent des neuroblastomes, qui sont d’origine sympathique.
Une translocation chromosomique t(11;22)(q24,q12) est retrouvée dans 88 à 95 % des sarcomes d’Ewing.
On a montré que le gène EWS, jusque-là inconnu, situé sur le chromosome 22, était fusionné en amont d’un oncogène connu, Fli1, situé sur le chromosome 11.
Dans la translocation t(21;22), le gène EWS est fusionné avec ERG, un oncogène appartenant à la même famille que Fli1, et dans la translocation t(7;22) avec ETV. Le gène EWS est impliqué dans tous les cas.
Le type de translocation semble, dans certains cas, avoir un effet sur le pronostic et sur le comportement biologique de la tumeur.
On peut rencontrer des translocations intéressant le gène EWS dans d’autres tumeurs, comme certaines tumeurs à cellules géantes, les tumeurs desmoplastiques intraabdominales ou le liposarcome et le chondrosarcome myxoïdes.
Des cellules murines (NIH3T3) transfectées avec un gène de fusion EWS-Fli1 deviennent tumorales, alors que le gène EWS ou Fli1 seul est sans effet.
Des expériences plus récentes confirment le rôle de ce gène de fusion dans la régulation de certains mécanismes de prolifération cellulaire et pourraient également ouvrir des possibilités thérapeutiques nouvelles.
Un des rôles de Fli1 pourrait être d’inhiber l’apoptose des cellules tumorales.
L’absence apparente d’antigène d’origine neurale a pendant un temps servi à différencier le sarcome d’Ewing des autres tumeurs neuroectodermiques.
En réalité, il y a un continuum entre sarcome d’Ewing typique, sarcome d’Ewing atypique et PNET.
Le sarcome d’Ewing est la plus indifférenciée des lésions de cette famille.
Localisation :
A – SUR LE SQUELETTE :
Le sarcome d’Ewing peut atteindre tous les os du squelette, avec une légère préférence pour les os longs, touchés dans 50 à 60 % des cas.
Le fémur est atteint dans environ 15 à 27 % de l’ensemble des cas, puis le tibia, l’humérus et le péroné.
Le membre inférieur est plus souvent atteint.
Lors d’atteinte des os plats, le bassin est le plus souvent concerné, avec 20 % de l’ensemble des cas, puis plus rarement les côtes (troisième localisation en fréquence), l’omoplate ou les vertèbres.
Des cas d’atteintes du pied, ou plus exceptionnellement de la main et du crâne, peuvent aussi se voir.
Dans les lésions des os du crâne, la mandibule est plus souvent atteinte que le maxillaire ou la base du crâne.
Des sarcomes d’Ewing des parties molles sans aucune connexion squelettique peuvent exister, ainsi que des sarcomes d’Ewing de la peau.
B – SUR L’OS :
Le sarcome d’Ewing est plus souvent localisé dans la zone diaphysaire des os longs que les autres tumeurs osseuses primitives, mais il atteint préférentiellement la zone métaphysaire et particulièrement diaphysométaphysaire.
Avant fermeture du cartilage de croissance, l’atteinte épiphysaire est exceptionnelle.
Étude clinique :
A – FORME HABITUELLE :
Le symptôme révélateur habituel du sarcome d’Ewing est la douleur, presque toujours présente.
Cette douleur est d’abord intermittente, puis devient persistante au repos, pouvant être d’intensité progressivement croissante sur plusieurs semaines.
Il n’est pas rare de voir des patients après plusieurs mois d’évolution.
Le deuxième symptôme pouvant motiver la consultation est l’existence d’une tuméfaction, de taille rapidement croissante, qui peut devenir très importante.
B – FORMES PARTICULIÈRES :
Du fait des multiples localisations possibles et de l’importance fréquente de l’envahissement des parties molles au cours de l’évolution des sarcomes d’Ewing, les circonstances de diagnostic sont extrêmement variables et trompeuses.
Les localisations pelviennes sont souvent muettes longtemps et le diagnostic est alors fait tardivement avec des lésions très volumineuses.
Des localisations pelviennes profondes peuvent entraîner une symptomatologie urologique par compression de la vessie, voire par retentissement sur les cavités hautes.
Des troubles vésicosphinctériens peuvent aussi être en rapport avec l’envahissement du plexus sacré. Les sarcomes d’Ewing du sacrum ou de la sacro-iliaque peuvent être révélés par une sciatalgie.
Les localisations rachidiennes peuvent se manifester par une attitude scoliotique dont le caractère raide et douloureux doit alerter.
Le premier symptôme peut aussi être une paraplégie.
Le sarcome d’Ewing est, avec le granulome éosinophile, l’une des plus fréquentes lésions tumorales des côtes chez l’enfant.
Elle peut se révéler de façon atypique par une symptomatologie respiratoire en rapport avec un épanchement pleural.
Habituellement, les formes costales se révèlent par une tuméfaction douloureuse.
Dans les formes à développement intrathoracique, il peut ne pas y avoir de tuméfaction palpable.
Le caractère inflammatoire de la tumeur, les douleurs, l’existence d’une fièvre, peuvent faire penser à une ostéomyélite ou une ostéoarthrite, d’autant qu’un syndrome inflammatoire biologique peut se voir. Une fracture pathologique est rarement révélatrice d’un sarcome d’Ewing, même si cela est possible.
Les circonstances de découverte des rares sarcomes d’Ewing des parties molles sont celles des tumeurs des tissus mous, et c’est seulement l’examen histologique qui peut faire le diagnostic.
Étude radiologique :
A – FORME HABITUELLE TYPIQUE :
1- Radiologie conventionnelle :
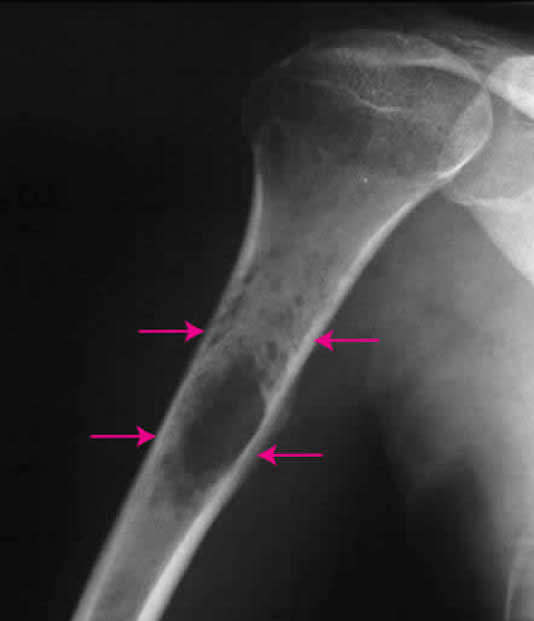
L’aspect radiologique du sarcome d’Ewing est variable, mais prend habituellement les caractéristiques d’une tumeur maligne agressive.
La forme typique (qui n’est pas la plus fréquente), siégeant sur les os longs, est celle d’une lésion lytique, entourée d’une réaction périostée fusiforme en fines lamelles parallèles au grand axe de l’os, réalisant une image en « bulbe d’oignon ».
La lyse osseuse est mal limitée, irrégulière, donnant un aspect vermoulu, avec des corticales apparaissant mouchetées.
L’éperon de Codman est inconstant.
Certaines lésions, en particulier métaphysaires, peuvent aussi avoir un aspect plutôt condensant, inhomogène, avec une réaction périostée souvent plurilamellaire et spiculée, irrégulière, pouvant être associée à une masse dans les parties molles.
Dans les lésions métaphysaires, le cartilage de croissance, quand il est ouvert, est habituellement respecté.
Tous les aspects peuvent se voir, mais un élément très caractéristique est le volume de la tumeur des parties molles, souvent radiotransparente, qui contraste avec une atteinte osseuse souvent discrète.
Cet aspect se voit souvent dans les lésions des os plats.
Dans les atteintes des côtes, les radiographies de gril costal sont parfois difficiles à lire, montrant mal la lyse osseuse, alors que la radiographie de thorax de face montre une masse arrondie ou ovalaire à développement intrathoracique, avec parfois un épanchement pleural associé.
2- Tomodensitométrie :
L’examen tomodensitométrique (TDM) n’est pas le meilleur examen dans les atteintes des os longs périphériques.
Il précise mal l’envahissement intramédullaire, ainsi que l’atteinte des parties molles dans lesquelles il n’y a habituellement pas de calcifications.
On lui préfère d’emblée l’imagerie par résonance magnétique (IRM), même si cette dernière donne de moins bonnes images de l’atteinte osseuse corticale.
La TDM est plus intéressante dans les lésions des côtes où la masse des parties molles contraste avec le parenchyme pulmonaire.
Lors des atteintes rachidiennes, la TDM va montrer l’existence d’une atteinte osseuse, souvent lytique, et l’éventuel envahissement intracanalaire.
Cet examen est parfois demandé dans le cadre du bilan d’une radiculalgie et il permet alors d’en rapporter la cause à une lésion tumorale et non discale.
Dans les lésions pelviennes, la TDM peut préciser une image douteuse en radiologie conventionnelle, en montrant un aspect spiculé, mité, irrégulier de la corticale osseuse et en montrant l’atteinte des parties molles.
3- Imagerie par résonance magnétique :
Dans les formes diaphysaires ou métaphysodiaphysaires, l’IRM montre en T1 une atteinte intramédullaire souvent plus importante que ne le laisserait supposer la radiologie conventionnelle.
En T2, il existe un renforcement hétérogène du signal de la masse tumorale qui permet de bien montrer l’atteinte des parties molles.
Lors de l’injection de gadolinium, la prise de contraste est habituellement intense.
Les images en T1, en haut contraste à saturation de graisse, donnent des images encore plus précises de l’atteinte des parties molles.
Le contraste entre l’importance de la masse des parties molles visible en IRM et une atteinte radiologique parfois discrète est assez caractéristique du sarcome d’Ewing.
4- Scintigraphie et artériographie :
En scintigraphie au technétium, le sarcome d’Ewing s’accompagne d’une hyperfixation du traceur radioactif, non spécifique.
Hormis le bilan d’extension, cet examen n’a d’intérêt que pour localiser une zone pathologique mal visible en radiologie conventionnelle et orienter d’autres examens.
L’artériographie, qui ne fait pas partie des examens à visée diagnostique, montre habituellement une hypervascularisation tumorale.
B – AUTRES FORMES RADIOLOGIQUES :
Au début, les lésions radiographiques sont parfois très discrètes et ne sont reconnues que rétrospectivement.
Dans ces cas, seules la scintigraphie, la TDM ou l’IRM permettent des diagnostics précoces.
À côté des atteintes diaphysaires centrales typiques, des formes diaphysaires corticales peuvent se voir, avec une lyse corticale excentrée, à type de saucérisation, et un développement prédominant dans les parties molles, sans extension intramédullaire.
Des formes purement condensantes peuvent rarement se voir, dans certaines localisations métaphysaires, ou parfois au bassin.
Les sarcomes d’Ewing des parties molles, sans atteinte osseuse, nécessitent d’emblée le recours à l’IRM, même si dans certains cas le premier examen demandé a été une échographie.
Bilan d’extension :
A – EXTENSION LOCALE :
Les radiographies conventionnelles et les radiomensurations sont surtout utiles à la planification chirurgicale et à préparer le matériel nécessaire.
Le meilleur examen pour évaluer l’extension locale est l’IRM qui donne des images précises dans les plans frontal et sagittal et permet de mesurer le volume tumoral.
Elle est actuellement indispensable et doit être faite avant la biopsie.
Comme dans toute tumeur maligne, l’IRM doit comprendre l’os atteint en entier, avec des mensurations précises de l’envahissement intramédullaire.
L’IRM précise au mieux l’envahissement dans les parties molles, sur les séquences en T2, ou en T1 et saturation de graisse (fat sat) et les rapports avec les principaux troncs nerveux et vasculaires.
L’IRM peut également montrer un envahissement intra-articulaire.
Les rapports de la tumeur et du cartilage de conjugaison sont bien visibles.
Cette IRM est répétée au cours de la chimiothérapie, car les modifications du volume tumoral donnent une idée de l’efficacité du traitement sur la lésion.
Une IRM préopératoire est indispensable, car parfois l’envahissement intraosseux peut avoir augmenté en cours de chimiothérapie.
Dans les localisations de la paroi thoracique, la TDM donne des renseignements comparables à l’IRM du fait du contraste offert par le poumon.
L’envahissement intraosseux est mal évalué par l’IRM, du fait de l’obliquité des côtes et des petites dimensions du canal médullaire.
Les localisations costales ont la particularité de donner des métastases pleurales et tout épanchement pleural doit être ponctionné à la recherche de cellules tumorales.
Il est également indispensable de rechercher une extension postérieure vers le canal rachidien au travers du trou de conjugaison.
Les localisations rachidiennes peuvent donner des métastases à tout le névraxe, dont l’exploration complète par IRM est indispensable.
Une ponction lombaire, si elle est possible, peut aider à rechercher un envahissement de l’axe cérébrorachidien.
Les reconstructions permises par la TDM sont parfois utiles pour imaginer la configuration tridimensionnelle de la tumeur.
L’artériographie paraît intéressante dans les localisations où une dissection vasculaire est nécessaire.
Elle n’est pas demandée par toutes les équipes chirurgicales, sauf avant reconstruction par greffon vascularisé.
Dans ce cas précis, l’artériographie est aussi nécessaire pour vérifier la présence d’une anatomie vasculaire normale du site donneur.
B – EXTENSION GÉNÉRALE :
Le bilan d’extension à distance comporte systématiquement une scintigraphie au technétium, à la recherche de métastases osseuses.
Des radiographies des zones fixant à la scintigraphie ou douloureuses, même en l’absence de fixation, sont nécessaires.
En cas de doute, une IRM est demandée, et si l’imagerie n’est pas suffisante, on peut faire des biopsies orientées pour préciser l’existence ou l’absence de métastases.
Le bilan d’extension doit comporter des radiographies de face et de profil ainsi qu’un examen TDM du thorax, à la recherche de localisations pulmonaires.
Le bilan comporte systématiquement une recherche d’extension à la moelle hématopoïétique, par biopsie au trocart et ponctions médullaires multiples.
La recherche de transcrit de fusion 11-22 ou 21-22 par reverse transcription-polymerase chain reaction (RT-PCR) sur un pool de ponctions médullaires permet d’affirmer l’existence d’une diffusion micrométastatique.
Anatomie pathologique :
Le sarcome d’Ewing appartient au groupe des tumeurs à petites cellules rondes, groupe constitué d’entités bien définies sur le plan biologique et évolutif mais aux caractéristiques histologiques proches.
A – BIOPSIE :
1- Gestion de la biopsie :
La biopsie doit être de volume suffisant pour permettre les analyses anatomopathologiques, bactériologiques, de biologie moléculaire (recherche de transcrit), cytogénétiques (caryotype).
La biopsie doit être rapidement acheminée au laboratoire, soit à l’état frais, soit dans un liquide conservateur de type RPMI, pour permettre une étude histologique de qualité ainsi que les analyses cytogénétiques et de biologie moléculaire.
À défaut, la biopsie est placée dans un liquide fixateur, mais les analyses biologiques (recherche de transcrit) ne sont plus possibles.
L’étalement sur lame de la biopsie permet en quelques minutes d’orienter vers une tumeur à petites cellules rondes, ou bien vers une infection, un granulome éosinophile, une tumeur à cellules géantes ou cartilagineuse.
Le diagnostic n’est porté définitivement qu’après analyse histologique.
La recherche du transcrit par RT-PCR ne nécessite qu’une très faible quantité de matériel.
Le pathologiste doit donc congeler systématiquement un fragment de chaque biopsie afin de pouvoir réaliser l’analyse en cas de nécessité.
2- Aspect microscopique :
Le sarcome d’Ewing est, dans sa forme classique, constitué de petites cellules tumorales agencées en larges travées séparées par un abondant tissu fibreux.
* Cytologie :
Les cellules sont identiques entre elles, arrondies, mesurant entre 12 et 14 µm.
Leur noyau, ovalaire ou arrondi, est pourvu d’une chromatine dense mais dispersée, et délimité par une fine membrane nucléaire.
L’activité mitotique est variable, souvent faible.
Les cytoplasmes sont pâles et très peu abondants, aux limites floues.
Ils comportent du glycogène, identifiable par les colorations complémentaires, acide périodique Schiff (PAS) et carmin de Best.
Des cellules plus sombres, appelées dark cells, ont été décrites comme le second type cellulaire du sarcome d’Ewing.
Il s’agit en fait de cellules tumorales altérées spontanément ou lors de la biopsie.
* Architecture :
Le plus souvent, les cellules forment de larges travées ou des plages de très forte densité en cellules tumorales, sans agencement particulier.
Il existe parfois des images de pseudorosette liées à des nécroses cellulaires, sans neurofibrille. Les cellules se regroupent parfois autour de petits capillaires.
Plus rarement, les cellules s’agencent en lobules, en alvéoles, ou constituent de fines travées séparées par de larges trousseaux de fibres de collagène, disposition appelée filigree pattern.
Cette disposition s’observe à la périphérie de la tumeur et correspond à l’infiltration du tissu musculoaponévrotique.
Le réseau de fibres de réticuline apparaît très peu abondant entre les cellules tumorales, alors qu’il est très dense autour des vaisseaux.
Les placards tumoraux sont par ailleurs entourés par un abondant tissu hyalin.
* Remaniements :
Ils sont extrêmement fréquents, liés à la fragilité des cellules, associant hémorragies et surtout nécrose de coagulation.
Leur importance peut être telle qu’ils rendent impossible un diagnostic de certitude.
S’observent aussi très fréquemment, en périphérie des plages tumorales, des cellules aux noyaux étirés ou écrasés, altérations généralement induites par le geste biopsique.
3- Analyse immunohistochimique :
Elle est décisive car l’aspect cytologique et la disposition architecturale ne sont pas toujours spécifiques.
Son rôle est double.
* Éliminer les diagnostics différentiels :
Pour une majorité d’entre eux, des marqueurs spécifiques sont disponibles, comme le CD 45 et les marqueurs T et B pour les lymphomes.
Toutefois, avec le développement de techniques immunohistochimiques plus sensibles et l’emploi du démasquage antigénique, des expressions aberrantes de ces marqueurs s’observent dans des sarcomes d’Ewing, comme la cytokératine ou la desmine.
Elles sont généralement focales et peu intenses.
* Apporter des éléments de diagnostic positif :
La protéine p30/32 codée par le gène Mic2 est exprimée en grande quantité dans les sarcomes d’Ewing et les PNET.
Son marquage est membranaire.
Son expression s’observe aussi dans un grand nombre de cellules normales de l’organisme, comme les fibroblastes, ainsi que dans différentes tumeurs comme certains carcinomes, etc.
Parmi les tumeurs à petites cellules rondes, les PNET sont l’une des seules à l’exprimer de façon intense et diffuse.
Toutefois, d’autres tumeurs à petites cellules ont été trouvées positives pour le Mic2, lymphome lymphoblastique, chondrosarcome mésenchymateux, ostéosarcome à petites cellules, rhabdomyosarcome alvéolaire et embryonnaire, mais aucune tumeur nerveuse ne semble l’exprimer.
Sa signification doit donc impérativement être corrélée à l’aspect histologique et aux données cliniques et radiologiques.
L’expression par la tumeur des marqueurs nerveux (NSE, neurofilaments, leu-7, etc) est plus rare.
De nouvelles techniques de recherche de la translocation et du transcrit de fusion à partir de tissus fixés et inclus en paraffine se développent et sont très prometteuses.
B – DIFFÉRENTES FORMES ANATOMOPATHOLOGIQUES :
1- Neuroépithéliome :
Il est considéré comme la forme la plus mature des lésions de la famille des PNET.
Son aspect microscopique est caractéristique, constitué des cellules de même morphologie mais qui se regroupent autour de lumières à centre fibrillaire, réalisant les rosettes d’Homer-Whright.
En immunohistochimie, elles expriment intensément les marqueurs nerveux (neuron specific enolase [NSE], neurofilaments, leu-7), la chromogranine et la synaptophysine, et de façon conjointe le Mic2. Tous les intermédiaires morphologiques existent entre le neuroépithéliome et le sarcome d’Ewing.
Pour certains, le diagnostic de PNET doit être réservé aux tumeurs comportant des images de rosettes typiques ou qui expriment au moins deux marqueurs nerveux en immunohistochimie.
2- Sarcome d’Ewing atypique :
Les cellules tumorales sont plus volumineuses, aux noyaux irréguliers en forme et en taille, à chromatine plus dense, à cytoplasme abondant et bien limité.
Cet aspect microscopique est trompeur.
Sa présentation clinique et son pronostic sont similaires à la forme typique.
L’étude immunohistochimique et l’analyse cytogénétique sont déterminantes.
3- Autres variantes :
D’autres variantes sont décrites, comme les formes rhabdoïdes, à grandes cellules et épithélioïdes.
Elles sont extrêmement rares, identifiées grâce à la biologie moléculaire car leur aspect histologique est particulièrement trompeur.
C – ASPECT MACROSCOPIQUE :
La tumeur possède des contours irréguliers et des limites très imprécises.
C’est une masse blanc grisâtre, molle, luisante, prenant dans les secteurs nécrotiques une consistance liquide, voire laiteuse.
Le sarcome d’Ewing infiltre diffusément la pièce osseuse, s’insinuant entre les travées ostéoïdes, pénétrant la corticale au travers des canaux d’Havers en détruisant peu ou pas les structures osseuses.
La réaction périostée s’effectue parallèlement au plan osseux, mais rapidement franchie par la tumeur, une nouvelle ossification se développe, réalisant l’aspect classique en « pelure d’oignon ».
L’infiltration des parties molles est extrêmement fréquente, la tumeur se propageant le long des insertions tendineuses et des aponévroses, s’étendant à distance de la lésion osseuse.
Cette infiltration explique la discordance parfois observée entre l’évaluation radiologique de la tumeur et l’infiltration plus importante observée sur la pièce de résection.
D – ÉTUDE DE LA PIÈCE DE RÉSECTION :
La technique de prise en charge est analogue à celle utilisée pour l’ostéosarcome, mais une grande attention doit être déployée pour évaluer la qualité d’exérèse. Deux caractéristiques du sarcome d’Ewing gênent parfois l’évaluation du pourcentage de cellules résiduelles viables.
Du fait de sa fragilité, il est difficile de déterminer si la nécrose observée est spontanée ou induite par les traitements.
Du fait qu’il altère de façon minime les tissus qu’il infiltre, il est difficile d’évaluer la surface de la tumeur avant tout traitement, à la différence de l’ostéosarcome qui laisse une marque souvent nette.
E – BIOLOGIE MOLÉCULAIRE ET RECHERCHE DU TRANSCRIT DE FUSION :
La recherche des transcrits de fusion par RT-PCR sert non seulement pour le diagnostic positif du sarcome d’Ewing, mais aussi au diagnostic d’extension métastatique par l’analyse des prélèvements de moelle osseuse.
La sensibilité importante de ces techniques permet de mettre en évidence la présence de cellules tumorales non détectées par la cytologie conventionnelle.
La recherche du gène de fusion EWS-Fli1 peut également se faire par hybridation in situ avec des sondes spécifiques fluorescentes (FISH).
Ces techniques de RT-PCR ou de FISH permettent un diagnostic rapide.
Diagnostic différentiel :
A – CLINIQUE :
L’existence de douleurs ostéoarticulaires associées à une tuméfaction inflammatoire et à une fièvre peut évoquer une ostéomyélite.
Le diagnostic différentiel peut être rendu encore plus difficile par l’existence d’un syndrome inflammatoire biologique et par l’aspect radiologique de certaines localisations de sarcome d’Ewing.
L’aspect macroscopique lors de la biopsie peut également être trompeur, avec une tumeur qui peut prendre un aspect pseudopurulent.
Il est donc indispensable, devant toute suspicion d’ostéomyélite, de faire un prélèvement anatomopathologique à la recherche d’une éventuelle tumeur maligne et en particulier le sarcome d’Ewing.
B – RADIOLOGIQUE :
L’image en « bulbe d’oignon », souvent décrite comme étant caractéristique du sarcome d’Ewing, peut se voir dans d’autres pathologies comme l’ostéomyélite ou les lymphomes non hodgkiniens.
Les lymphomes osseux primitifs peuvent également prendre un aspect moucheté et vermoulu comme le sarcome d’Ewing.
Les lymphomes touchent habituellement des patients plus âgés que le sarcome d’Ewing.
Le diagnostic est fait par l’étude histologique et immunohistochimique.
Les métastases osseuses de neuroblastome sont une des premières causes de lésions osseuses lytiques de l’enfant de moins de 5 ans.
Les métastases osseuses, souvent multiples, peuvent révéler la maladie.
Le dosage sanguin et urinaire des catécholamines confirme le diagnostic et la scintigraphie permet de localiser la tumeur primitive.
Les métastases osseuses d’autres tumeurs viscérales se voient surtout chez l’adulte.
L’aspect radiologique de certains sarcomes d’Ewing peut évoquer à tort un ostéosarcome, quand la réaction périostée se fait avec des spicules perpendiculaires à la direction de la corticale ou dans les localisations métaphysaires condensantes.
Le granulome éosinophile, ou histiocytose langerhansienne, donne parfois des lésions lytiques d’évolution très rapide pouvant faire discuter un sarcome d’Ewing.
La présence de multiples localisations osseuses, avec des images de lyse à l’emporte-pièce au crâne, oriente vers une histiocytose langerhansienne.
Dans certains cas de lésion unique, seule la biopsie permet d’orienter le diagnostic.
Le kyste osseux anévrismal peut prendre un aspect très agressif, avec une lyse importante et une volumineuse extension dans les parties molles.
L’existence de niveaux liquides, si elle évoque fortement le kyste anévrismal, n’est pas pathognomonique.
La biopsie redresse le diagnostic.
C – DIAGNOSTIC DIFFÉRENTIEL ANATOMOPATHOLOGIQUE :
Une lésion à petites cellules rondes soulève une grande variété de diagnostics différentiels que le tableau clinique et radiologique permet de limiter.
1- Chez l’enfant :
Chez le jeune enfant avant 5 ans, avant d’envisager un sarcome d’Ewing, deux diagnostics, le lymphome et le neuroblastome, doivent être systématiquement évoqués.
Lymphome et leucose sont très proches morphologiquement mais leur profil immunohistochimique permet de les reconnaître aisément.
Le neuroblastome est alors une métastase osseuse révélatrice.
La tumeur comporte généralement des rosettes et entre les cellules un fond neurofibrillaire. L’ostéosarcome à petites cellules, qui est rare, s’observe chez l’adolescent et l’adulte jeune.
L’ostéoformation tumorale, qui permet de porter le diagnostic, est parfois très peu abondante, pas toujours intéressée par la biopsie et doit être distinguée de l’ostéoformation réactionnelle, parfois très abondante.
Comme la tumeur exprime aussi le Mic2 et que des observations avec la translocation 11-22 ont été rapportées, le diagnostic de cette tumeur peut être problématique.
La tumeur desmoplasique à petites cellules rondes est une tumeur de l’adolescent ou du jeune adulte, généralement abdominale, qui peut envahir l’os par contiguïté ou par dissémination métastatique.
Elle est généralement plus hétérogène que le sarcome d’Ewing, avec une expression en immunohistochimie de multiples marqueurs, vimentine, cytokératine, marqueurs neuroendocrines, musculaires, etc.
2- Chez l’adulte :
La rareté du sarcome d’Ewing après 30 ans doit faire envisager les autres tumeurs à petites cellules rondes, en particulier lymphome et métastases.
Les métastases osseuses de carcinome peuvent simuler morphologiquement un sarcome d’Ewing dans deux cas :
– la tumeur de Merkel est un carcinome neuroendocrine cutané constitué de cellules de petite taille, identifiable grâce à l’expression en immunohistochimie de la cytokératine et des marqueurs neuroendocrines sous forme de boules intracytoplasmiques ;
– le carcinome à petites cellules, dont l’origine est souvent de siège bronchique et dont les cellules sont fragiles, souvent écrasées, voire nécrosées.
L’immunohistochimie associe l’expression des marqueurs neuroendocrines et de la cytokératine.
Deux autres diagnostics difficiles, qui ont une présentation clinique de tumeur osseuse primitive, peuvent aussi s’observer chez l’adulte jeune : l’ostéosarcome à petites cellules déjà cité et le chondrosarcome mésenchymateux, qui peut aussi siéger dans les parties molles.
Le contingent cartilagineux, souvent minoritaire, réalise des nodules disposés à l’emporte-pièce dans le contingent à petites cellules.
L’absence de marqueur spécifique et la faible abondance du contingent cartilagineux en font un diagnostic difficile à porter.
Orientation thérapeutique :
A – MÉTHODES DE TRAITEMENT :
Un protocole européen commun de traitement des sarcomes d’Ewing et tumeurs apparentées existe depuis 1999, mais reste encore en cours d’évaluation.
Ce protocole, appelé Euro-Ewing 99 pour European Ewing tumour Working Initiative of National Groups, est identique pour les enfants et pour les adultes.
Le traitement jusqu’à présent utilisé chez l’enfant, en France, était le protocole Ewing 93 de la Société française d’oncologie pédiatrique.
Chez l’adulte, certains utilisent aussi le protocole EICESS 92 (European Intergroup Cooperative Ewing’s Sarcoma Study).
Dans tous les cas, le traitement actuel du sarcome d’Ewing comprend un traitement général, la chimiothérapie, et un traitement local, chirurgie et éventuellement radiothérapie.
1- Traitement local : radiothérapie
Dès 1921, Ewing avait insisté sur la grande radiosensibilité de la tumeur qu’il décrivait.
La radiothérapie est restée longtemps le traitement local de choix du sarcome d’Ewing, mais laissait de lourdes séquelles que les techniques modernes tentent de limiter.
Le champ doit inclure toute la tumeur, le trajet de la biopsie, ou la voie d’abord chirurgicale, mais doit éviter d’irradier des tissus sains.
Les marges de sécurité sont réduites à 2 cm.
Les rayons X de haute énergie, qui ont une meilleure pénétration, sont plus volontiers utilisés que les rayonnements classiques.
L’irradiation doit préserver les épiphyses, ce qui diminue son retentissement sur la croissance chez l’enfant. Lors des irradiations pelviennes, on peut transposer chirurgicalement les ovaires et implanter des prothèses pour écarter la vessie ou les intestins.
Chez le garçon, la protection des testicules est systématique. Les complications aiguës ou chroniques peuvent être potentialisées par l’administration concomitante d’agents radiosensibilisants, comme l’actinomycine.
L’irradiation thoracique peut entraîner une fibrose pulmonaire et retentit sur la croissance du poumon et de la cage thoracique. Elle peut également intéresser le coeur et potentialise l’effet cardiotoxique de la doxorubicine.
Éviter les irradiations circonférencielles aux membres permet de ne plus rencontrer l’effet de garrot.
La radiothérapie peut encore, dans certains cas de tumeurs rapidement évolutives ou inopérables, rester le seul traitement local.
Parfois, la radiothérapie peut être associée à la chirurgie pour essayer de diminuer le risque de récidive locale.
Certaines équipes utilisent la radiothérapie, avant ou pendant la chirurgie, pour diminuer le volume tumoral et limiter le champ d’irradiation au seul volume tumoral.
L’inconvénient de ces techniques est de faire appel systématiquement à une irradiation.
Actuellement, la radiothérapie est plus souvent utilisée en postopératoire et seulement si elle est nécessaire.
En cas de radiothérapie postopératoire, la dose recommandée dans le protocole Euro-Ewing 99 est de 44 à 54 Gy.
Pour les patients non opérés, la dose recommandée est de 54 Gy, avec surimpression jusqu’à 60 ou 64 Gy en fonction de l’âge et de la localisation.
Au rachis, la dose maximale tolérable par la moelle épinière est de 45 Gy.
L’existence d’un épanchement pleural tumoral dans les lésions de la paroi thoracique ou d’une atteinte du liquide céphalorachidien est une indication d’irradiation de l’hémithorax ou de l’ensemble du névraxe.
La radiothérapie est administrée 2 à 4 semaines après la chirurgie en cas de chimiothérapie conventionnelle, et 8 à 10 semaines en cas de chimiothérapie à hautes doses.
2- Traitement local : chirurgie
Grâce aux progrès de la chimiothérapie, la chirurgie est redevenue de nos jours le traitement local de référence du sarcome d’Ewing.
Avec l’apparition des premières chimiothérapies, et grâce à l’effet de la radiothérapie, le traitement était devenu de moins en moins chirurgical, car il consistait alors essentiellement en l’amputation.
Les complications de la radiothérapie, le contrôle local incomplet offert par l’irradiation et les bons résultats des résections ont conduit à modifier les indications.
Progressivement, les publications ont montré une amélioration du pronostic grâce au recours à la chirurgie conservatrice dans le traitement local par rapport aux résultats obtenus avec les radiothérapies seules.
Actuellement, il est admis que la chirurgie doit être conservatrice, tout en répondant aux impératifs de la chirurgie carcinologique, car le pronostic est plus général que local.
Les très rares indications d’amputation concernent certaines localisations des extrémités distales et les tumeurs des très jeunes enfants (pour certains auteurs).
Les tumeurs impossibles à réséquer du fait des séquelles fonctionnelles prévisibles ou pour des raisons carcinologiques sont généralement des indications d’irradiation.
La reconstruction des localisations métaphysaires avec atteinte épiphysaire, au genou ou à l’humérus et au fémur proximal, se fait par prothèse massive.
La seule particularité de ces reconstructions est le jeune âge des patients qui peut nécessiter le recours à des prothèses de croissance, télescopiques, destinées à compenser le sacrifice du cartilage de croissance.
Dans les lésions des os longs, le sarcome d’Ewing est souvent purement diaphysaire ou métaphysodiaphysaire, permettant la conservation des épiphyses et des cartilages de croissance.
La reconstruction fait alors appel soit à un segment d’allogreffe diaphysaire, soit de préférence à un greffon de péroné vascularisé que certaines équipes associent à une allogreffe. Les résultats de ces reconstructions par péroné vascularisé sont spectaculaires chez l’enfant.
La résection du péroné proximal peut nécessiter le sacrifice du nerf fibulaire commun, imposant alors le transfert secondaire du tendon tibial postérieur pour compenser la paralysie des releveurs du pied.
En cas de résection diaphysaire, il faut parfois stabiliser la malléole externe par vis.
Au pied ou à la main, des résections carcinologiques sont parfois possibles, mais l’amputation est souvent préférée. Dans les lésions costales, les limites précises de l’envahissement intraosseux sont difficiles à mesurer sur l’imagerie.
La résection doit donc intéresser toute la côte qui est désarticulée du rachis en arrière et du cartilage en avant, avec l’espace intercostal sus- et sous-jacent.
La reconstruction consiste dans le rapprochement des deux côtes voisines avec fermeture simple des plans sus-jacents. Parfois, on fait appel à des plaques de tissu résorbable, voire à des lambeaux musculaires de rotation.
Dans les lésions postérieures de la côte, la résection emporte l’apophyse transverse en bloc avec la côte, et parfois même une partie du corps vertébral.
Les procédés de reconstruction, dans les localisations pelviennes ou rachidiennes, n’ont pas de particularité.
Le principal problème est d’évaluer précisément avant et pendant l’intervention les limites de la résection.
Au rachis, une résection large, en bloc, est rarement possible du fait de la présence du canal vertébral et de la moelle ou du sac dural et des racines, rendant une irradiation souvent nécessaire.
Les lésions de l’omoplate sont traitées par scapulectomie et la reconstruction peut se faire par simple suspension de l’humérus à la clavicule.
Les sarcomes d’Ewing du crâne et du massif maxillofacial peuvent rarement bénéficier d’une résection large, hormis les lésions de la mandibule qui sont accessibles à un traitement chirurgical avec reconstruction par greffe, de péroné vascularisé par exemple.
Les sarcomes d’Ewing des parties molles doivent être traités comme ceux d’autres localisations, par résection large.
3- Traitement général de la maladie : chimiothérapie
L’action du cyclophosphamide (Endoxant), de l’actinomycine (Dactinomycinet) et de la vincristine (Oncovint) contre le sarcome d’Ewing est connue depuis le début des années 1960.
La chimiothérapie permet de diminuer le volume de la tumeur primitive, ce qui en rend le traitement local plus facile et plus efficace, et permet de diminuer le développement de métastases à distance.
Depuis le milieu des années 1980, différents protocoles associant vincristine + actinomycine D + cyclophosphamide (VAC), plus ou moins doxorubicine (VACA), étoposide (VP-16, vépésidet) ou ifosfamide (Holoxant), ont permis d’améliorer le pronostic.
Dans les formes métastatiques au départ ou répondant mal à la chimiothérapie conventionnelle, des protocoles faisant appel à des chimiothérapies à hautes doses ont donné certains résultats encourageants.
Le protocole Ewing 93 comporte, pour les formes localisées, une chimiothérapie d’induction composée de trois à cinq cures initiales, (Adriblastinet-Adriamycinet) dites « Memphis », associant cyclophosphamide et doxorubicine, et parfois une association d’étoposide et d’ifosfamide.
La chimiothérapie d’entretien après chirurgie comporte, en fonction de la réponse de la tumeur , d’abord six cures de vincristine associée à de l’actinomycine D, puis dans une seconde phase, six cures de « Memphis » répétées toutes les 3 semaines.
Dans les autres cas, la chimiothérapie d’entretien comporte six cures d’étoposide ifosfamide au lieu des six cures de « Memphis ». Le protocole Euro-Ewing 99 comporte une induction par six cures de vincristine + ifosfamide + doxorubicine + étoposide (VIDE) à des intervalles de 3 semaines.
Après chirurgie ou bilan radioclinique dans les formes non chirurgicales, la chimiothérapie fait ensuite appel à différentes drogues, soit vincristine + actinomycine + ifosfamide (VAI), soit vincristine + actinomycine + cyclophosphamide (VAC).
Dans les formes de mauvais pronostic, les deux protocoles font appel à une chimiothérapie à hautes doses associant busulfan et melphalan.
La cure est suivie de la réinjection de cellules souches périphériques prélevées auparavant par cytaphérèse et congelées.
B – TRAITEMENT DES MÉTASTASES :
1- Traitement général :
Le traitement des formes métastatiques comporte un traitement général et un traitement local des métastases, en plus de celui de la tumeur primitive.
Le traitement général fait bien entendu appel aux chimiothérapies déjà décrites.
Certaines équipes ont également fait appel à des irradiations du corps en entier ou de l’hémicorps, après prélèvement de cellules souches périphériques, ou autogreffe de moelle hématopoïétique.
Ces irradiations n’ont pas apporté de résultats durables et sont actuellement souvent abandonnées.
2- Traitement local :
Dans les métastases pulmonaires, le traitement peut comporter soit une résection chirurgicale des nodules visibles en imagerie, soit des lobectomies.
Du fait de la haute radiosensibilité du sarcome d’Ewing à la radiothérapie, les lésions secondaires pulmonaires peuvent être traitées par irradiation.
L’irradiation pulmonaire nécessite la prévention des pneumopathies interstitielles à Pneumocystis carinii.
Elle est contre-indiquée après chimiothérapie à hautes doses par bulfalan.
Quand les métastases osseuses sont uniques, on peut dans certains cas discuter un traitement par résection chirurgicale, comme pour la tumeur primitive.
La plupart des métastases osseuses sont irradiées.
C – INDICATIONS ACTUELLES DES DIFFÉRENTS TRAITEMENTS :
Selon le protocole Ewing 93, dans les formes localisées opérables, le traitement local repose sur la chirurgie qui est effectuée après cinq cures de « Memphis » et un nouveau bilan d’extension.
L’analyse de la pièce de résection permet d’évaluer la réponse histologique à la chimiothérapie et la qualité des marges de résection.
Les bons répondeurs, ayant moins de 5 % de cellules viables, reçoivent six cures de vincristine + actinomycine D, puis six cures de « Memphis ».
En cas de résection incomplète, une irradiation complémentaire est effectuée.
Les patients ayant entre 5 et 30 % de cellules viables reçoivent une radiothérapie postopératoire puis six cures de vincristine + actinomycine D, suivies de six cures d’étoposide et holoxan.
Les patients ayant au moins 30 % de cellules résiduelles reçoivent deux cures de VP-16 + holoxan, puis une chimiothérapie à hautes doses par misulban-melphalan, et une greffe de cellules souches périphériques, ainsi qu’une radiothérapie locale.
Le traitement des patients non opérables dépend de la taille de la tumeur initiale et repose sur les mêmes principes.
Dans les formes métastatiques, le traitement initial reste le même.
En cas de réponse apparente supérieure à 50 %, et si les métastases semblent opérables, les patients reçoivent deux cures d’étoposide (VP-16) + ifosfamide (holoxant), puis une chimiothérapie à hautes doses et une greffe de cellules souches.
Si la tumeur initiale est opérable, elle est traitée chirurgicalement ; sinon, elle est irradiée.
Dans les autres cas, les indications sont discutées individuellement.
Dans le protocole Euro-Ewing 99, après la résection, on utilise, chez les bons répondeurs, sept cures, soit de VAI, soit de VAC.
Chez les mauvais répondeurs ayant plus de 10 % de cellules résiduelles, le protocole compare sept cures de VAI versus une chimiothérapie à hautes doses par busulfan-melphalan.
Dans les formes métastatiques, aux poumons ou à la plèvre, tous les patients reçoivent six cures de VIDE, puis on décide de la poursuite du traitement local et général de consolidation en fonction de la réponse à la chimiothérapie évaluée cliniquement et par l’imagerie, et parfois par biopsie de la tumeur.
Le moment du traitement local dépend des modalités du traitement général choisi.
En cas de chimiothérapie conventionnelle ou de tumeur facilement opérable, la résection chirurgicale est faite après la sixième cure de VIDE, puis la chimiothérapie est poursuivie selon la randomisation.
Dans les cas où une chimiothérapie à hautes doses a été choisie, si la chirurgie est complexe et risque d’entraîner un retard dans la poursuite de la chimiothérapie, le traitement local est repoussé à la fin de la chimiothérapie.
En fonction de la réponse histologique à la chimiothérapie et du caractère complet ou non de l’exérèse, une radiothérapie complémentaire peut être indiquée.
Les patients dont la tumeur continue d’évoluer localement (qu’ils aient des métastases ou non) ou dont les métastases ne répondent pas au traitement par VIDE peuvent recevoir soit une chimiothérapie à hautes doses avec busulfan et melphalan, soit une irradiation de la tumeur primitive et des sites métastatiques suivie d’une chimiothérapie de consolidation par double-ME.
Évolution. Pronostic. Résultats des traitements :
A – ÉVOLUTION :
Le sarcome d’Ewing est une lésion d’évolution habituellement rapide.
Localement, elle a tendance à envahir les parties molles, de façon parfois très importante, et elle peut aussi s’étendre à l’ensemble de la cavité médullaire de l’os atteint.
Cet envahissement se fait au travers des canaux de Havers de l’os, puis par une véritable infiltration des tissus sains environnants.
Il y a quelques cas où la tumeur a, au contraire, une évolution assez lente, restant longtemps confinée dans l’os et ne donnant pas rapidement de métastases.
Cela explique probablement que le retard au diagnostic ne soit pas régulièrement retrouvé comme facteur de mauvais pronostic dans les études cliniques.
Le sarcome d’Ewing a une très forte tendance à donner des métastases par voie hématogène, ce qui en fait le pronostic.
Selon différentes études, environ 20 à 30 % des patients sont porteurs de métastases au diagnostic.
La survenue secondaire de métastases fait la gravité de la maladie, même dans les formes localisées au diagnostic.
Chez les patients sans métastases pulmonaires ni osseuses détectables, les méthodes de biologie moléculaire peuvent permettre de détecter des cellules tumorales dans la moelle osseuse.
Les principaux sites de diffusion métastatique sont le poumon (38 %), l’os (31 %) et la moelle hématopoïétique (11 %), ainsi que le névraxe dans les localisations rachidiennes ou la plèvre dans les localisations costales.
Dans le cas des métastases pleurales des tumeurs costales et des métastases au névraxe des localisations rachidiennes, la diffusion se fait par contiguïté et circulation des cellules dans la cavité pleurale ou le liquide céphalorachidien.
Le rachis est un des sites préférentiels des métastases osseuses hématogènes.
On peut également voir des métastases intradurales dans 5 % des cas environ.
L’envahissement lymphatique est assez rare, présent dans moins de 10 % des cas.
B – PRONOSTIC :
Avant le développement de la chimiothérapie et malgré un traitement chirurgical radical ou une irradiation, moins de 10 % des patients survivaient, à cause des métastases qui ne manquaient pas de se développer.
Le premier facteur pronostique dans le sarcome d’Ewing est l’existence de métastases au diagnostic.
La probabilité de survie sans récidive à 5 ans, qui va jusqu’à 70 % dans les séries modernes en l’absence de métastases, tombe alors à moins de 30 %.
Le pronostic des patients ayant des métastases osseuses est très mauvais, nettement inférieur à celui des patients ayant des métastases seulement pulmonaires.
La détection de cellules tumorales dans le sang circulant par RT-PCR n’a pas d’influence sur le pronostic, contrairement à la positivité de la RTPCR sur les biopsies médullaires.
Les tumeurs de grande taille, de volume tumoral initial supérieur à 100, voire 200 mL, ou une plus grande dimension supérieure à 8 cm, semblent de moins bon pronostic.
À traitement local équivalent et à réponse histologique égale, ce facteur perd de son influence.
Il semble également que les tumeurs les plus volumineuses soient aussi celles qui ont le plus de métastases, ce qui peut introduire un biais dans certaines études.
Généralement, les tumeurs des côtes ou des membres sont de meilleur pronostic que celles du bassin, du rachis et de l’omoplate.
L’influence de la localisation n’est pas retrouvée avec la même intensité dans les études portant sur les tumeurs non métastatiques, et Carrie rapporte des survies sans récidive à 5 ans de 72 % chez des patients opérés pour sarcome d’Ewing du bassin.
Ces différences sont en partie dues au fait que les tumeurs du bassin sont plus souvent métastatiques au diagnostic, plus volumineuses et que leur traitement local est plus difficile.
Pour certains, les sarcomes d’Ewing des tissus mous seraient de mauvais pronostic, ce qui est également controversé.
L’existence d’un syndrome inflammatoire biologique franc, d’une anémie, d’une fièvre élevée au diagnostic, ou une élévation de la lacticodéshydrogénase (LDH) sont des facteurs péjoratifs.
Les résultats sont habituellement moins bons chez les sujets les plus âgés.
Il semble néanmoins que l’âge des patients ait un rôle moins significatif quand les protocoles de traitement sont équivalents chez les enfants et les adultes et que les localisations sont comparables.
Par exemple, Bacci a montré dans un travail récent que le pronostic des patients âgés de plus de 40 ans était peu différent de celui des patients plus jeunes, avec respectivement 53 % contre 58 % de survie sans récidive à 5 ans.
L’existence d’une fracture pathologique n’est pas en soi un facteur de mauvais pronostic si le traitement est correctement conduit.
Le rôle du degré de différenciation histologique de la tumeur est encore controversé actuellement, ainsi que le type de translocation. Le facteur essentiel du pronostic, après traitement, est la réponse histologique à la chimiothérapie.
Par exemple, pour Bacci, la probabilité de survie sans récidive à 5 ans était de 28 % chez les mauvais répondeurs contre 77 % chez les bons répondeurs.
Le délai avant récidive était plus long, en moyenne 51 mois chez les bons répondeurs contre 32 chez les mauvais.
Dans l’étude rétrospective conjointe SFOP-EICESS, le taux de survie sans récidive à 5 ans des patients ayant moins de 10 % de cellules viables après chimiothérapie était de 73 % contre 45 % pour les patients ayant au moins 10 % de cellules résiduelles.
Environ 30 % des tumeurs avaient une mauvaise réponse à la chimiothérapie.
Les tumeurs chirurgicalement inextirpables ont un moins bon pronostic.
Les récidives très tardives, au-delà de 10 ans, ne sont pas exceptionnelles dans le sarcome d’Ewing et nécessitent une surveillance prolongée, et les études rapportant des résultats à seulement 5 ans de recul demandent à être confirmées sur de plus longs reculs.
Les récidives locales sont plus fréquentes après radiothérapie isolée qu’après chirurgie seule ou association de chirurgie et de radiothérapie.
Après radiothérapie isolée, le taux de contrôle local est estimé à 70 à 80 % dans les lésions périphériques et 60 % pour les lésions proximales.
Tous les auteurs s’accordent sur la nécessité d’avoir des marges de résection larges.
Par exemple, selon Ozaki, après chirurgie seule, le taux de récidive locale est de moins de 10 %, et si les marges de résection sont larges, ce taux de récidive est d’environ 5 % contre 12 % en cas de marges contaminées.
L’adjonction d’une radiothérapie à la chirurgie permet d’améliorer le contrôle local mais dans des proportions souvent peu significatives.
Le pronostic des récidives locales (habituellement accompagnées de diffusion métastatique) est très mauvais, avec moins de 10 % de survie à 5 ans, malgré les traitements par chimiothérapie à hautes doses avec greffe de moelle.
Les récidives précoces ont encore plus mauvais pronostic que celles survenant après 2 ans.
Le pronostic des récidives métastatiques reste mauvais, inférieur à 10 % de survie, sans seconde récidive à 5 ans.
Il est moins péjoratif en cas de métastase pulmonaire unique qui peut être accessible à un traitement chirurgical, et en cas de métastase tardive que de métastase précoce.
En cas de récidive après une chimiothérapie conventionnelle, le recours à une chimiothérapie à hautes doses permettra peut-être d’améliorer le pronostic à long terme.
C – RÉSULTATS DES TRAITEMENTS ACTUELS :
Nous avons déjà vu qu’avec les traitements récents, dont les résultats peuvent être connus à 5 ou 10 ans de recul, le pronostic du sarcome d’Ewing était de 65 à 70 % de survie sans récidive à 5 ans dans les formes non métastatiques au départ, et d’environ 30 % de survie sans récidive à 5 ans dans les formes métastatiques au départ.
D – COMPLICATIONS À DISTANCE DU TRAITEMENT :
Les résultats à long terme dépendent aussi de la survenue de complications directement liées aux traitements.
Un certain nombre de décès sont imputables à la chimiothérapie, en particulier défaillances cardiaques par cardiomyopathie progressive liée à la doxorubicine.
La surveillance tardive régulière de la fonction cardiaque est nécessaire, en particulier au moment d’interventions chirurgicales ou de grossesses.
Le retentissement sur la fertilité de la chimiothérapie est inconstant.
Habituellement, la fertilité n’est pas modifiée par les chimiothérapies conventionnelles à base d’anthracyclines, de vincristine ou d’actinomycine, alors que les chimiothérapies à hautes doses entraînent régulièrement une stérilité.
Chez le garçon pubère, une cryopréservation de sperme doit être proposée.
Une complication du traitement chirurgical des tumeurs des côtes est l’apparition secondaire d’une scoliose, convexe du côté de la résection.
Ces scolioses sont évolutives et entraînent un retentissement respiratoire d’autant plus important qu’une irradiation a été associée.
Les autres complications tardives de la chirurgie n’ont rien de spécifique.
Les complications de l’irradiation, en particulier viscérales, ont déjà été évoquées.
Certaines de ces complications sont désormais évitées. D’autres complications sont plus particulièrement orthopédiques.
L’irradiation d’un cartilage de croissance va entraîner chez l’enfant une stérilisation au moins partielle de celui-ci et son retentissement est d’autant plus important que l’enfant est jeune.
Les traitements de ces inégalités sont difficiles.
On peut recourir à des allongements ou à l’épiphysiodèse du côté sain.
La correction des déviations angulaires peut se faire par épiphysiodèse, avant la fin de la croissance, ou par ostéotomie.
Les fractures en os irradié posent des problèmes liés à la sclérose des parties molles avoisinantes et à la mauvaise consolidation de l’os irradié.
La fracture est parfois le mode de révélation d’un sarcome radio-induit et une biopsie est toujours nécessaire lors du traitement d’une telle fracture. L’incidence des sarcomes radio-induits est d’environ 6 à 10%, 20 ans après traitement du sarcome d’Ewing.
Ces lésions secondaires radio-induites peuvent survenir très tardivement et leur incidence élevée dans le sarcome d’Ewing paraît en partie liée aux chimiothérapies associées et à des facteurs génétiques.
Conclusion :
Le sarcome d’Ewing reste une maladie relativement rare.
Son diagnostic repose actuellement sur l’histologie et sur les données de la biologie moléculaire.
Son pronostic a été significativement amélioré grâce aux thérapeutiques modernes.
Son traitement repose sur l’association d’une chimiothérapie entourant la résection chirurgicale complète de la tumeur.
La radiothérapie doit rester réservée à quelques indications particulières en raison de ses complications.
Le but des traitements actuels est de diminuer les séquelles directement liées aux thérapeutiques dans les formes dites de bon pronostic et d’améliorer les résultats du traitement des formes dites de mauvais pronostic, dont les taux de survie restent actuellement décevants.

")





")



{kind=link}