



















Introduction :
Les LCP non épidermotropes d’origine T lymphocytaire constituent un groupe hétérogène.
Ils sont classés selon la taille des cellules lymphomateuses, en LCP à petites et moyennes cellules, et en LCP à grandes cellules.
 Parmi les LCP à grandes cellules, l’expression par les cellules tumorales de l’antigène d’activation CD30 a permis d’individualiser le groupe des LCP CD30+ caractérisé, à la différence des LCP à grandes cellules n’exprimant pas cet antigène, par un pronostic favorable, et ce quelle que soit leur morphologie, anaplasique, immunoblastique ou pléomorphe.
Parmi les LCP à grandes cellules, l’expression par les cellules tumorales de l’antigène d’activation CD30 a permis d’individualiser le groupe des LCP CD30+ caractérisé, à la différence des LCP à grandes cellules n’exprimant pas cet antigène, par un pronostic favorable, et ce quelle que soit leur morphologie, anaplasique, immunoblastique ou pléomorphe.
Ils sont classés dans la classification des lymphomes cutanés proposée par l’organisation européenne de recherche sur le traitement des cancers (EORTC), parmi les lymphomes « indolents », aux côtés de la PLy dont ils sont proches.
Le spectre des lymphoproliférations CD30+ est un spectre continu comprenant les LCP CD30+, les PLy de type A, ainsi que des formes frontières entre ces deux entités.
Ces lymphoproliférations cutanées ont comme caractéristique commune d’exprimer l’antigène CD30, mais ont aussi de nombreuses similitudes anatomocliniques et évolutives et peuvent parfois s’associer ou évoluer l’une vers l’autre.
D’autre part, elles ont de nombreuses ressemblances anatomocliniques avec les lymphomes ganglionnaires CD30+, mais dont il paraît important de les distinguer, car leur évolutivité différente appelle un traitement particulier et peut suggérer une pathogénie distincte.
Antigène CD30 :
L’antigène CD30 a été identifié grâce à l’anticorps monoclonal Ki-1 développé contre les cellules de Reed-Sternberg qui caractérisent la MH, puis son expression par les cellules anaplasiques de lymphomes ganglionnaires a été mise en évidence, permettant leur identification.
Le CD30 est un récepteur transmembranaire de la famille du récepteur du tumor necrosis factor (TNF).
Son gène est localisé en 1p36, près de ceux des autres membres de la famille du TNF qui interviennent dans la différenciation et la prolifération des lymphocytes.
Le ligand de l’antigène CD30 (CD30L) est une cytokine proche du TNF dont les effets pléiotropes sur des lignées lymphocytaires CD30+ pourraient ouvrir des perspectives pour une immunothérapie spécifique.
Cependant, le CD30L a un effet différent sur les lignées cellulaires dérivées de MH où il induit la prolifération cellulaire, alors qu’il induit l’apoptose dans les lignées de cellules lymphomateuses anaplasiques.
Le CD30s, forme soluble du CD30, est détectable dans le sérum des malades porteurs d’une MH ou d’un lymphome ganglionnaire CD30+.
Son taux y est corrélé au stade de la maladie, et surtout à un mauvais pronostic et à une résistance au traitement.
À l’inverse, son taux diminue avec la réponse au traitement. Sa valeur n’a jusqu’alors pas été étudiée dans les LCP CD30+.
Lymphomes ganglionnaires anaplasiques CD30+ :
A – INTRODUCTION :
La reconnaissance de l’antigène CD30 a révélé que des tumeurs anciennement considérées comme des « histiocytoses malignes » étaient des lymphomes anaplasiques.
L’anticorps monoclonal Ki-1, qui avait initialement permis de les individualiser, a ensuite été supplanté par l’anticorps monoclonal Ber-H2 reconnaissant un épitope du CD30 résistant à la fixation, permettant donc l’étude de larges séries et de prélèvements rétrospectifs, et la description des caractéristiques de ces lymphomes.
B – CLINIQUE :
Ces lymphomes, relativement rares (5 à 8 % des lymphomes ganglionnaires), ont une distribution bimodale.
Ils sont plus fréquents chez l’enfant et l’adulte jeune, avec un premier pic vers 20 ans, mais il existe aussi un deuxième pic vers 70 ans.
Leur présentation est agressive, surtout chez le jeune où plus de 50 % des malades sont d’emblée en stade III à IV avec des signes généraux, une polyadénopathie et une atteinte extraganglionnaire, surtout cutanée (20 à 30 %) et osseuse.
L’atteinte médullaire est rare, mais quand elle existe, elle est de mauvais pronostic.
C – HISTOPATHOLOGIE :
La prolifération tumorale est diffuse à prédominance intrasinusale détruisant l’architecture ganglionnaire.
L’infiltrat est pléomorphe, avec de grandes cellules ressemblant aux cellules de Reed-Sternberg présentes dans la MH.
Les différents variants histologiques sont rassemblés dans un même groupe.
Dans la majorité des cas, les cellules lymphomateuses sont de phénotype T ou nul. Des lymphomes ganglionnaires CD30+ de phénotype B sont aussi décrits, surtout lors du sida, mais se rapprochent des lymphomes B diffus à grandes cellules.
Les cellules lymphomateuses, par définition CD30+, expriment des antigènes d’activation comme l’antigène épithélial de membrane (EMA) (65 à 83 %), le BNH9 (reconnaît les antigènes des groupes sanguins H et Y) (60 %), le CBF78 (nouvel antigène des lymphocytes T) (80 %), le CD45 (antigène commun des leucocytes) (45 %) et plus rarement le CD15 (LeuM1) (inférieur à 20 %).
Ce profil phénotypique permet le diagnostic différentiel avec la MH, en particulier dans sa forme riche en cellules tumorales.
Ces lymphomes ganglionnaires CD30+ ne sont que rarement (10 à 20 %) associés au virus d’Epstein-Barr (EBV).
D – GÉNÉTIQUE :
Un réarrangement clonal du gène de la chaîne ç du récepteur des lymphocytes T est détecté en polymerase chain reaction (PCR) dans plus de 60 % des cas.
La translocation t(2;5) consiste en la fusion des gènes de la nucléophosmine (NPM) et de l’anaplastic lymphoma kinase (ALK).
Récemment, le clonage du point de cassure a permis de disposer d’amorces pour détecter les transcrits NPMALK par RT-PCR et les points de cassure chromosomiques par PCR.
L’anticorps ALK1 a secondairement été mis au point pour l’immunodétection de la protéine ALK exprimée par les lymphocytes porteurs de la t(2;5).
Il a ainsi été montré que la t(2;5) est un événement moléculaire caractéristique de ces lymphomes et que sa détection y constitue un outil non seulement diagnostique mais aussi pronostique.
En effet, bien que le pouvoir oncogénique de NPM-ALK ait été démontré in vivo et in vitro, la t(2;5) est paradoxalement associée dans ces lymphomes à un bon pronostic.
E – ÉVOLUTION ET TRAITEMENT :
Ils nécessitent des chimiothérapies agressives, voire des greffes médullaires, mais réagissent souvent bien à ces traitements malgré leur présentation clinique agressive et indépendamment du stade d’extension initial, et leur pronostic est moins mauvais que celui des autres lymphomes T ganglionnaires à grandes cellules.
En fait, le pronostic dépend de l’âge des malades, avec une survie à 5 ans à 54 % chez les malades âgés, alors qu’elle est de 77 % chez les enfants et les adultes jeunes.
Lymphomes cutanés primitifs T CD30+ :
A – CARACTÉRISTIQUES :
Ils représentent 9 % des LCP dans le recensement européen de Willemze.
Alors que les lymphomes cutanés CD30+ faisant suite à un mycosis fongoïde (MF) et qui correspondent alors à des « MF transformés » sont de mauvais pronostic, les LCP CD30+, sans antécédent de lymphoprolifération préexistante, sont caractérisés par une survie prolongée.
Le caractère cutané primitif est défini par l’absence d’extension extracutanée pendant au moins 6 mois après le diagnostic initial.
Ce bon pronostic a permis, au cours de ces 10 dernières années, leur individualisation, les opposant aux lymphomes ganglionnaires CD30+, même si ces derniers ont un meilleur pronostic que les autres lymphomes T ganglionnaires à grandes cellules.
Il est donc important pour le choix thérapeutique de distinguer ces LCP CD30+ des localisations cutanées secondaires ou simultanées d’un lymphome systémique CD30+.
Ce problème de diagnostic différentiel est fréquent, car la peau est la première des localisations extraganglionnaires des lymphomes ganglionnaires CD30+ T et les localisations cutanées secondaires, à type de nodules souvent sous-cutanés, peuvent régresser spontanément comme des lésions de LCP CD30+.
Leur définition repose sur les critères établis par Beljaards :
– 1. présence sur la biopsie cutanée initiale de plus de 75 % de cellules lymphomateuses exprimant l’antigène CD30 ;
– 2. absence de lésions de type PLy ;
– 3. absence d’antécédent de PLy, de MF ou d’autre lymphome ;
– 4. absence d’atteinte extracutanée au moment du diagnostic.
Toutefois, le critère d’exclusion concernant l’existence de lésions de type PLy ou d’antécédents de PLy paraît discutable.
En effet, le pronostic des PLy associées à des LCP CD30+ est bon, à la différence des MF secondairement transformés en lymphomes à grandes cellules CD30+ et on peut alors se demander où classer les LCP CD30+ faisant suite à une PLy.
B – CLINIQUE :
Les LCP CD30+ surviennent le plus souvent chez l’adulte de plus de 50 ans, plus fréquemment de sexe masculin (sex-ratio : 3/2).
Il n’y a donc pas de distribution bimodale et ils sont exceptionnels chez l’enfant, contrairement aux lymphomes ganglionnaires CD30+.
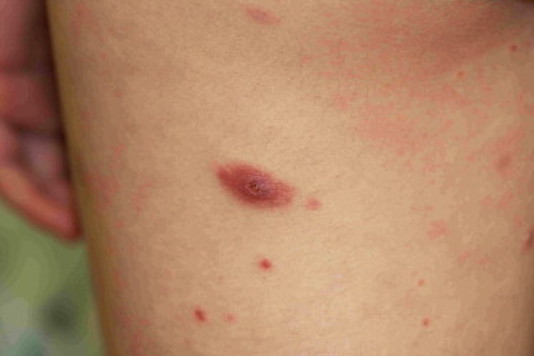
Ils se présentent sous la forme de lésions papulonodulaires ou tumorales de 2 à 10 cm de diamètre, le plus souvent localisées ou régionales, plus rarement multicentriques, volontiers ulcérées et nécrotiques.
Des tumeurs parfois très volumineuses sont aussi observées.
Elles sont de croissance rapide et sont surtout caractérisées par des épisodes de régression spontanée.
Toutefois, ces épisodes ne sont pas spécifiques des LCP puisqu’ils peuvent aussi être observés dans des localisations cutanées de lymphomes ganglionnaires CD30+.
D’autre part, ils peuvent, dans les LCP, être suivis par une évolution défavorable.
Enfin, des LCP CD30+ se révélant par une érythrodermie ont aussi été décrits.
C – HISTOPATHOLOGIE :
L’infiltrat lymphomateux est non épidermotrope, mais on observe souvent une hyperplasie pseudoépithéliomateuse en regard de l’infiltrat.
Les grandes cellules lymphomateuses sont groupées en nappes denses.
Elles sont soit de type anaplasique (80 % des cas) avec un noyau rond ou ovale, irrégulier, un ou plusieurs nucléoles éosinophiles et un large cytoplasme, soit de type immunoblastique ou pléomorphe.
Toutefois, la morphologie des cellules lymphomateuses n’influence pas le pronostic et ces LCP T CD30+ anaplasiques, pléomorphes ou immunoblastiques, sont rassemblés dans un même groupe.
Il existe aussi quelques cellules de type Reed-Sternberg éparses. Par définition, plus de 75 % des cellules tumorales expriment l’antigène CD30, avec un marquage membranaire et golgien.
Elles sont le plus souvent CD3+, CD4+.
L’expression d’antigènes d’activation tels que l’EMA, le CD15, est discutée.
Ainsi, si pour certains l’expression d’EMA est restreinte aux lymphomes anaplasiques ganglionnaires et constitue un caractère distinctif avec les LCP CD30+, d’autres études observent une expression de l’EMA dans 33 % à 41 % des cas, sans que cette expression n’ait de valeur pronostique.
Il existe aussi fréquemment une expression anormale de l’oncoprotéine p53 et une expression de marqueurs de cytotoxicité tels que le granzyme B et l’antigène intracellulaire des lymphocytes T (TiA1), comme dans les lymphomes anaplasiques ganglionnaires.
Enfin, ces LCP CD30+ ne sont habituellement pas associés à la présence de protéines ou de transcrits de l’EBV.
D – GÉNÉTIQUE :
L’étude génotypique de ces LCP montre le plus souvent un réarrangement clonal du gène de la chaîne ç récepteur T au niveau des prélèvements cutanés.
En revanche, peu de données sont disponibles à propos de la clonalité des lymphocytes circulants.
La présence de transcrits NPM-ALK et d’une expression de la protéine ALK, liée à la translocation t(2;5), constitue un événement très rare dans les LCP CD30+, ce qui suggère que la t(2;5) n’est vraisemblablement pas impliquée dans leur pathogenèse et constitue donc un argument en faveur d’un comportement biologique différent de celui des lymphomes systémiques CD30+.
L’existence de très rares LCP t(2;5)+ implique toutefois que la détection de la t(2;5) n’est pas utilisable à l’échelon individuel pour distinguer LCP et localisations cutanées secondaires d’un lymphome ganglionnaire, même si l’expression d’ALK est significativement plus fréquente dans les localisations secondaires.
D’autre part, des techniques trop sensibles comme la RT-PCR nichée sont à éviter pour le diagnostic car la détection de transcrits isolés peut ne refléter que la présence de cellules « bystander », comme cela a été décrit pour d’autres translocations au niveau ganglionnaire.
E – ÉVOLUTION ET TRAITEMENT :
Le bon pronostic des LCP CD30+, quelle que soit leur morphologie, avait été soupçonné par de petites séries et a été confirmé par les 47 malades de Beljaards où la survie à 5 ans est de 90 % et la médiane de survie de 52 mois, puis par deux séries de 86 et 65 malades.
Cependant, une extension extracutanée peut survenir tardivement, après 6 mois, dans 25 % des cas.
Bien que leur signification n’ait pas été étudiée statistiquement, des caractéristiques cliniques ont pour certains une valeur pronostique défavorable, telles que des lésions cutanées multicentriques initiales, alors que d’autres études ont ensuite montré que ce caractère multicentrique n’avait pas de valeur pronostique statistiquement défavorable.
Seuls les épisodes de régression spontanée et l’âge inférieur à 60 ans ont été démontrés comme statistiquement corrélés à un pronostic favorable.
De même, aucun marqueur phénotypique, y compris l’expression de l’EMA ou de p53, n’a de valeur prédictive pour le pronostic.
Depuis que leur bon pronostic a été reconnu, le traitement des LCP CD30+ ne repose pas sur des chimiothérapies agressives qui ne permettent pas d’obtenir de meilleure réponse et doivent être réservées à des malades avec une extension extracutanée secondaire.
La chirurgie ou la radiothérapie sont indiquées pour des lésions locorégionales.
Le méthotrexate à la dose de 15 à 25 mg par semaine, voire l’interféron-alpha-2a, pourraient être intéressants, de par leur bonne tolérance et leur efficacité, pour des lésions multicentriques, même si cela n’a pas été validé de façon prospective.
Toutefois, quel que soit le traitement entrepris, de fréquentes récidives cutanées sont observées.
Même si elles ne sont pas forcément associées à un mauvais pronostic, elles retentissent sur la qualité de vie des malades et il serait intéressant d’évaluer leur impact afin d’y adapter le choix thérapeutique.
F – LYMPHOMES CUTANÉS CD30+ ET INFECTION PAR LE VIRUS DE L’IMMUNODÉFICIENCE HUMAINE :
Les malades infectés par le virus de l’immunodéficience humaine (VIH) ont un risque accru de développer des lymphomes systémiques B agressifs. Les lymphomes cutanés sont eux rarement décrits au cours du sida.
Contrairement aux lymphomes systémiques-sida, ces lymphomes cutanés ont un phénotype T prédominant, et la fréquence des lymphomes cutanés CD30+ est remarquable (Beylot-Barry M, Vergier B, Masquetier B, Bagot M, Joly P, Souteyrand P et al. The spectum of cutaneous lymphonas in human immunodeficiency virus infection : a study of 21 cases. Am J Surg Pathol, 1999 [sous press]) et est inhabituelle par rapport à celle des LCP CD30+ de l’immunocompétent.
Les interactions entre le VIH et le CD30L récemment évoquées pourraient expliquer la fréquence de lymphomes T CD30+ au cours de l’infection par le VIH, mais n’expliquent pas pourquoi ils sont fréquents surtout au niveau cutané et non au niveau ganglionnaire. Ces LCP CD30+ surviennent chez des malades très immunodéprimés (Beylot- Barry et al. 1999, sous presse).
Leur présentation clinique est proche des LCP CD30+ de l’immunocompétent, avec des nodules ou des tumeurs souvent localisés, de fréquents épisodes de régression spontanée, et le décès n’est qu’exceptionnellement dû au lymphome mais est lié à l’immunodépression, ce qui amène à proposer d’éviter des traitements agressifs risquant de majorer l’immunodépression.
L’absence d’expression de la protéine ALK les rapproche également des LCP CD30+ de l’immunocompétent (Beylot-Barry et al. 1999, sous presse). En revanche, des transcrits EBER sont souvent détectés dans ces lymphomes (Beylot-Barry et al. 1999, sous presse), les rapprochant alors des lymphomes systémiques B CD30+ associés au sida.
La présence de l’EBV est souvent parallèle à l’hyperexpression de l’oncoprotéine p53, ce qui peut suggérer une interaction EBV-p53.
Enfin, ces lymphomes ne sont pas associés à la présence du virus herpès humain de type 8 (Beylot-Barry et al. 1999, sous presse).
Papulose lymphomatoïde :
A – INTRODUCTION :
La PLy de type A appartient au spectre des lymphoproliférations CD30+ et a de nombreuses similitudes anatomocliniques avec les LCP CD30+.
B – CLINIQUE :
La moyenne d’âge des sujets atteints est légèrement inférieure à celle des LCP CD30+ car les cas pédiatriques sont plus fréquents. Elle se caractérise par la survenue chronique d’éruptions autorégressives faites de lésions multiples de différents types en fonction de leur âge.
La lésion initiale est une papule érythémateuse, parfois un peu brunâtre, qui va évoluer spontanément en quelques jours vers la nécrose, puis vers la régression spontanée en quelques semaines, laissant une cicatrice dyschromique atrophique.
Plus rarement, il peut exister des nodules ou des plaques.
Les lésions sont nombreuses, d’une dizaine à une centaine, disséminées sur le corps, en particulier sur le tronc et les membres alors que le visage est rarement atteint.
C – HISTOPATHOLOGIE :
Les lésions de PLy ont une histologie variable, non seulement selon l’âge des lésions mais aussi selon le type de la PLy.
Le type A, qui appartient au spectre des lymphoproliférations cutanées CD30+, est caractérisé par un infiltrat non épidermotrope formant un triangle bien limité pointant vers la profondeur du derme, à grand côté épidermique.
Il est fait de plusieurs îlots périvasculaires de grands lymphocytes d’allure anaplasique de phénotype T CD3+, CD4+, CD30+, mêlés à un infiltrat polymorphe de petits lymphocytes, d’histiocytes, de neutrophiles et d’éosinophiles.
D – GÉNÉTIQUE :
La détection par PCR d’une population monoclonale T est fréquente dans la PLy (69 %) dans une proportion proche de celle des LCP, sans qu’elle soit prédictive d’un pronostic défavorable.
Comme dans les LCP CD30+, l’étude de la clonalité des lymphocytes circulants a rarement été réalisée, et les rares résultats décrits ne permettent pas de conclure quant à sa prévalence et à sa signification.
Enfin, la t(2;5) ne paraît pas impliquée dans leur pathogénie.
E – ÉVOLUTION ET TRAITEMENT :
Les PLy peuvent succéder ou précéder un lymphome cutané ou ganglionnaire CD30+, une MH ou un MF, dans un pourcentage de cas qui était estimé de l’ordre de 10 à 20 % d’après des séries limitées ou des cas ponctuels.
En fait ce pourcentage serait plus faible, de l’ordre de 5 %, selon la série de 70 PLy de Beljaards, qui recense aussi 39 cas de la littérature de PLy associées à des lymphoproliférations.
Dans cette série, le risque d’avoir une PLy associée à un lymphome paraît plus important si la PLy est riche en grandes cellules CD30+.
Les malades sont plutôt de sexe masculin, et sont plus âgés que ceux avec une PLy isolée (59 ans versus 42 ans).
L’ancienneté, la localisation, la taille et l’étendue des lésions de PLy ne paraissent pas être des facteurs prédictifs de l’évolution.
L’association PLy-MF est la plus fréquente des associations (38 % des PLy associées à des lymphoproliférations cutanées ou ganglionnaires) et a un bon pronostic ne modifiant pas l’évolution naturelle de la PLy ou du MF.
De même, l’association PLy-lymphome cutané CD30+ (12 % des PLy associées à un lymphome) paraît de bon pronostic.
En revanche, l’association à un lymphome ganglionnaire anaplasique CD30+ (20 %) ou à une MH (24 % des associations) sont de plus mauvais pronostic.
Le traitement des PLy n’est pas toujours satisfaisant en termes d’efficacité et s’adapte à la gêne entraînée par les lésions.
Si elles sont peu importantes, l’abstention thérapeutique est préférable, sinon le traitement peut aller de la PUVAthérapie aux moutardes azotées, jusqu’au méthotrexate ou à l’interféron-alpha-2a.
F – DIAGNOSTIC DIFFÉRENTIEL :
Le diagnostic différentiel entre PLy et LCP CD30+ n’est pas toujours aisé et il existe même d’authentiques formes frontières entre PLy et LCP CD30+ ou formes « borderline » où la clinique et l’histologie sont discordantes.
Ainsi, il peut s’agir d’une tumeur unique évoquant cliniquement un LCP CD30+ mais ayant une histologie de PLy ; Willemze considère alors le malade comme ayant un LCP CD30+.
Il peut, à l’inverse, s’agir d’une éruption de multiples papules évoluant vers la régression, évoquant cliniquement une PLy mais dont l’histologie est celle d’un LCP CD30+ ; il s’agit alors d’une PLy de type C, dont le pronostic paraît semblable à celui d’une PLy classique.
Les PLy de type B ne font quant à elles pas partie du spectre des lymphoproliférations CD30+, puisqu’alors que la clinique est la même que celle d’une PLy type A, l’histologie se rapproche plus d’un MF en plaques avec des cellules lymphoïdes de taille moyenne au noyau cérébriforme.
Toutefois, chez un même malade, des lésions de type PLy type A et PLy type B peuvent coexister ou se succéder.
Maladie de Hodgkin cutanée :
A – INTRODUCTION :
La MH est une pathologie ganglionnaire maligne caractérisée par la présence de cellules particulières, les cellules de Reed-Sternberg, dont l’origine est discutée (origine lymphoïde B, folliculaire dendritique ?).
Il s’agit de cellules de grande taille, avec fréquemment deux noyaux symétriques en miroir renfermant un volumineux nucléole. Bien que non spécifique de la MH, leur présence est indispensable pour porter le diagnostic, mais elles représentent moins de 5 % de l’infiltrat observé dans les ganglions hodgkiniens.
Les autres cellules sont des cellules de Hodgkin, des cellules lacunaires, ou encore des cellules anaplasiques.
Alors que les lésions cutanées les plus fréquentes au cours de la MH sont aspécifiques à type d’ichtyose, les lésions cutanées spécifiques sont rares, de l’ordre de 0,5 à 7,5 % des cas.
Il s’agit dans la quasitotalité des cas de lésions cutanées secondaires à une MH ganglionnaire connue, qui surviennent de façon tardive et qui signent un pronostic défavorable.
Toutefois, depuis les progrès thérapeutiques dans la MH, ces localisations cutanées secondaires sont plus rarement rapportées.
D’autre part, l’insuffisance des données phénotypiques dans les cas anciennement publiés ne permet pas toujours de porter formellement le diagnostic et d’exclure en particulier un LCP CD30+ ou une PLy.
Enfin, d’authentiques MH cutanées primitives ont exceptionnellement été décrites, et leur évolution indolente tend à les intégrer au spectre des lymphoproliférations cutanées CD30+.
B – CLINIQUE :
La présentation clinique des localisations secondaires de MH est peu spécifique et se rapproche de celle de LCP CD30+ ou de localisations cutanées secondaires de lymphomes ganglionnaires CD30+ avec des papules rouge-brun, des plaques, des nodules ou des tumeurs fréquemment ulcérées.
Ces lésions, souvent multiples, sont toutefois plus volontiers localisées dans le territoire de drainage du ganglion atteint, par extension lymphatique rétrograde.
Beaucoup plus rarement, il peut s’agir de l’extension directe au contact du ganglion atteint.
La dissémination hématogène est exceptionnelle.
Les observations suffisamment documentées de MH cutanées primitives sont au nombre de six.
Il s’agit aussi de nodules ulcérés ou de papules de nombre variable. Des épisodes de régression spontanée sont signalés dans deux des cinq cas de Sioutos et al, rappelant la présentation clinique d’une PLy.
Dans trois cas, l’atteinte cutanée reste isolée avec un recul de 20 ans, dans trois autres cas les lésions cutanées précèdent l’atteinte ganglionnaire de 2 mois à 6 ans, et le décès survient alors rapidement dans un cas où est soulevée la question de localisations cutanées d’une MH ganglionnaire encore occulte et non de MH cutanée véritablement primitive.
C – HISTOPATHOLOGIE :
Dans les localisations cutanées secondaires, comme dans les rares MH cutanées primitives, l’infiltrat nodulaire ou diffus envahit la totalité du derme.
Dans cet infiltrat polymorphe, les petits lymphocytes prédominent et sont mêlés à des éosinophiles et à des macrophages, alors que les cellules de Hodgkin, les cellules de Reed- Sternberg bi- ou multinuclées, et les cellules lacunaires sont éparses.
Les cellules de Reed-Sternberg expriment l’antigène CD30 et sont négatives pour le CD45, alors qu’elles ont un marquage membranaire pour le CD15, même si ce marquage n’est pas toujours observé dans la peau.
D – DIAGNOSTIC DIFFÉRENTIEL :
Celui-ci se pose surtout avec un LCP CD30+ ou une PLy qui peuvent aussi succéder ou précéder une MH, et beaucoup de cas anciennement publiés sont en réalité des lymphomes anaplasiques, ce qui a même fait discuter la réalité de MH primitivement cutanées.
S’il semble bien que cette entité existe, elle est toutefois extrêmement rare, et le diagnostic de PLy ou de LCP CD30+ doit être évoqué. Le polymorphisme de l’infiltrat, et surtout le phénotype CD30+/CD15+ membranaire/CD45- est en faveur d’une « MH cutanée », alors que les LCP CD30+ et les PLy sont CD30+/CD15 ± cytoplasmique/ CD45+, même si le marquage avec l’anti-CD15 est parfois négatif dans la peau.
D’autres marqueurs, tels que le BNH9 et le CBF78, non étudiés jusqu’ici dans les MH cutanées, pourraient être utiles dans le diagnostic différentiel.
E – GÉNÉTIQUE :
Peu de données sont disponibles au niveau cutané. Dans un cas, aucun réarrangement clonal B ou T n’est détecté par PCR, comme cela est d’ailleurs habituel dans la MH.
La translocation t(2;5) n’a pas été recherchée dans les MH cutanées, mais il a été montré qu’elle n’était pas impliquée dans les MH classiques.
F – ÉVOLUTION ET TRAITEMENT :
La rareté des MH cutanées primitives ne permet pas de conclure sur son pronostic, et son traitement repose soit sur l’abstention en raison de possibles régressions spontanées, soit sur la radiothérapie ou l’excision chirurgicale en première intention.
Pour les localisations cutanées secondaires, elles constituent un événement tardif de pronostic rapidement défavorable et leur traitement rejoint celui de la MH elle-même.
Une pathogénie commune ?
Malgré leurs différences pronostiques, la parenté entre leurs caractères cliniques, histologiques phénotypiques et moléculaires suggèrent une pathogénie commune entre lymphomes ganglionnaires anaplasiques CD30+, LCP CD30+ et PLy, d’autant que ces lymphoproliférations peuvent s’associer ou se succéder chez un même malade et qu’il a été montré chez des malades présentant une PLy associée à un lymphome que ces lymphoproliférations dérivaient d’un clone T commun.
Les mécanismes physiopathologiques conduisant à la transformation d’une PLy en un LCP CD30+ restent inconnus.
L’activité du TGF(transforming growth factor)-bêta qui inhibe la prolifération cellulaire, pourrait induire les phénomènes de régression des proliférations lymphocytaires CD30+.
À l’inverse, la perte de sensibilité au TGFbêta de lignées lymphocytaires issues de lymphomes CD30+ évolués, avec perte des récepteurs I et II du TGF-bêta, pourrait expliquer la disparition des phénomènes de régression et par là l’évolutivité clinique du lymphome.
La valeur pronostique de ces données, mises en évidence sur lignées cellulaires, reste à valider in vivo, en étudiant l’expression des récepteurs au TGF-bêta directement à l’échelon tissulaire (hybridation in situ, Northern blot…).
Le rôle de l’EBV, qui avait été évoqué dans les poussées de PLy, semble devoir être récusé d’après les études négatives en hybridation in situ et en immunohistochimie dans les PLy et les LCP CD30+, à l’exception des cas associés au sida.
La translocation t(2;5) qui est un événement moléculaire présent dans la majorité des lymphomes systémiques CD30+ paraît exceptionnelle dans les lymphoproliférations cutanées primitives CD30+ et ne joue donc certainement pas de rôle clé dans leur pathogenèse.
Perspectives :
Il n’y a pas actuellement de caractéristiques clinique, histopathologique, phénotypique ou moléculaire permettant de différencier une PLy d’un LCP CD30+, ni de critère diagnostique formel pouvant différencier une lymphoprolifération cutanée primitive CD30+ et une localisation cutanée d’un lymphome systémique CD30+ non encore connu lors du diagnostic.
Il n’existe pas non plus de facteur prédictif de l’évolutivité d’une PLy vers un lymphome.
Même si cette évolution demeure relativement rare, la surveillance à long terme de ces malades s’impose, une transformation étant suspectée devant l’augmentation de volume des lésions cutanées, l’absence de régression spontanée, et bien sûr l’apparition d’adénopathies.
Dans les lymphoproliférations cutanées primitives CD30+, il reste donc à identifier des événements moléculaires récurrents qui pourraient être utiles au diagnostic, voire au pronostic, et expliquer le comportement particulier de ces lymphoproliférations.
La recherche de gènes pouvant contribuer au développement des lésions par leur expression dans la tumeur ou son microenvironnement, et l’étude du profil d’expression génique de la tumeur dans des phases évolutives différentes (progression, régression, absence de régression) pourraient ainsi être développées.
D’autre part, l’intérêt de la détection du CD30s dans le sérum des malades pour le suivi évolutif et la prédiction du pronostic au cours des LCP CD30+ est à évaluer, ainsi que la fréquence et la signification d’un réarrangement monoclonal du gène de la chaîne ç du récepteur T dans les lymphocytes sanguins de ces malades, par comparaison à la peau.
Les LCP CD30+ sont définis par leur localisation purement cutanée.
Un passage des cellules lymphomateuses dans le sang circulant, dont le développement ne pourrait se faire que dans le microenvironnement cutané, pourrait peut-être expliquer la fréquence des récidives cutanées observées chez ces malades.







")



{kind=link}