



















Définition :
La dysplasie ostéofibreuse des os longs est un hamartome bénin.
Elle est presque toujours localisée au tibia et au péroné, se caractérise par une prolifération de tissu ostéofibreux anormal et peut s’accompagner de diverses déformations squelettiques.
Historique :
 Le premier cas décrit dans la littérature fait état d’une lésion tibiale évoquant une dysplasie ostéofibreuse des os longs : il a été publié par Frenghenheim en 1921, qui a nommé la maladie « ostéite fibreuse congénitale ».
Le premier cas décrit dans la littérature fait état d’une lésion tibiale évoquant une dysplasie ostéofibreuse des os longs : il a été publié par Frenghenheim en 1921, qui a nommé la maladie « ostéite fibreuse congénitale ».
Plus tard, sous divers synonymes, on retrouve dans la littérature des lésions similaires, interprétées de façon erronée comme étant une expression de la « dysplasie fibreuse » par certains auteurs, ou comme étant des « pseudarthroses congénitales du tibia » dans d’autres cas.
En 1966, Kempson décrit deux cas de lésions tibiales qu’il nomme « fibrome ossifiant des os longs », tout en précisant des aspects histologiques qui les distinguaient de la dysplasie fibreuse.
La première définition de la « dysplasie ostéofibreuse des os longs » est due à M Campanacci qui, en 1976, décrit la lésion comme étant une entité clinique distincte, avec des caractéristiques radiographiques spécifiques.
Les observations de l’auteur sur 22 cas font ressortir les différences entre la dysplasie ostéofibreuse et la dysplasie fibreuse, en termes d’âge et de site de survenue, d’aspects radiographique et histologique, ainsi que d’évolution.
Épidémiologie :
A – FRÉQUENCE :
La dysplasie ostéofibreuse est une lésion rare, dont l’incidence est légèrement supérieure dans le sexe masculin.
Elle survient surtout dans les 10 premières années de la vie, apparaissant souvent avant l’âge de 5 ans, bien qu’elle ait été décrite en de rares occasions à l’adolescence et dans les premières années de l’âge adulte.
B – LOCALISATIONS :
La lésion est quasi exclusivement localisée à la diaphyse tibiale et, dans 10 % des cas, le péroné homolatéral est touché simultanément.
Une localisation bilatérale aux deux tibias et aux deux péronés est exceptionnelle.
Dans quelques cas, elle n’a été observée que sur le péroné, voire sur d’autres os longs tels que le radius, le cubitus et l’humérus.
Un cas à localisations multiples avec implication simultanée des deux tibias, du cubitus et du péroné droit a été rapporté, bien que l’histologie n’ait pu être prouvée que pour une seule des lésions.
En général, la lésion est localisée au tiers moyen de la diaphyse tibiale, avec extension vers le tiers proximal et le tiers distal.
Il arrive, moins souvent et par ordre de fréquence, que le tiers distal et le tiers proximal de la diaphyse tibiale soient atteints isolément.
Lorsque la lésion concerne le péroné, elle touche toujours le tiers distal de la diaphyse.
Symptômes :
La lésion est indolore et souvent asymptomatique, à l’exception de la présence éventuelle d’une déformation squelettique.
Il existe habituellement une soufflure de l’os cortical qui entraîne volontiers une incurvation antérieure ou en varus-valgus de la diaphyse.
La lésion peut devenir douloureuse suite à une fracture (de fatigue ou pathologique), qui se voit dans 25 % des cas.
Néanmoins, ces fractures ont une évolution favorable et se consolident généralement très bien par un traitement orthopédique.
Une pseudarthrose serrée a parfois été observée, et un seul cas de pseudarthrose lâche a été rapporté.
Imagerie :
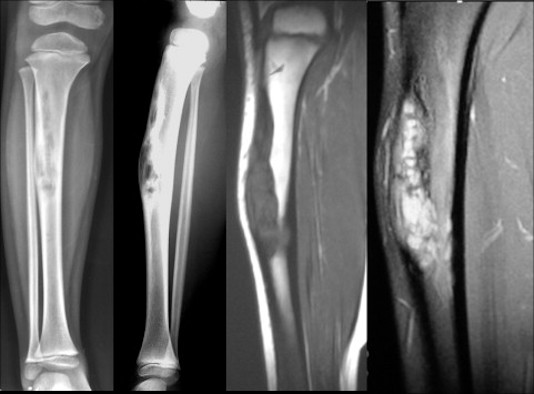
L’aspect radiographique est extrêmement évocateur et suffit souvent à établir le diagnostic au cours des 10 premières années de la vie.
La lésion se présente souvent comme une ostéolyse intracorticale excentrée, généralement associée à une déformation de type incurvation antérieure ou incurvation en varus-valgus de la diaphyse.
La face superficielle de la corticale est soufflée et amincie, disparaissant même en certains endroits, tandis que sa face profonde est délimitée par un liseré de sclérose qui peut parfois devenir très épais.
Conséquences de ces déformations, le canal médullaire est souvent le siège d’un rétrécissement, voire d’une fermeture complète, et l’ostéolyse peut parfois prendre un aspect multiloculaire.
On observe aussi, dans certains cas, une opacité en « verre dépoli » ressemblant à une dysplasie fibreuse au sein de l’ostéolyse ou, dans d’autres cas, certaines zones ayant tendance à s’ossifier.
Au tibia, la lésion est couramment excentrée, tandis qu’au péroné elle peut concerner toute la largeur de l’os.
Une tomodensitométrie confirme les observations radiographiques, montrant une lésion ostéolytique excentrée contenant un tissu solide.
La corticale externe apparaît soufflée et amincie, et peut même aller jusqu’à ne plus exister.
Le canal médullaire est rétréci et la lésion délimitée par une bordure d’ostéosclérose.
L’examen par résonance magnétique nucléaire (IRM) montre un faible signal homogène dans les images pondérées en T1.
L’intensité du signal augmente de façon non homogène dans les dernières images pondérées en T2.
L’examen par scintigraphie osseuse fait apparaître, au début de la phase dynamique, une légère captation préférentiellement au temps veineux.
À la phase terminale, une forte captation des nucléides radioactifs est observée au même niveau.
Histologie :
A – MACROSCOPIQUEMENT :
Le périoste est bien préservé et la corticale sous-jacente est très amincie.
Le tissu intratumoral est solide, adhérant aux parois de la cavité, variable dans sa couleur (blanc, jaune, rouge) et sa consistance (molle ou fibreuse).
B – HISTOLOGIQUEMENT :
On note un tissu fibreux prédominant avec des cellules moins nombreuses que dans la dysplasie fibreuse, présentant parfois un aspect fusiforme ressemblant au fibrome histiocytaire.
Ce tissu fibreux englobe de petites travées d’os réticulaire, toujours entourées d’ostéoblastes actifs, cette caractéristique typique aidant à distinguer la dysplasie ostéofibreuse de la dysplasie fibreuse.
Un aspect particulier de la dysplasie ostéofibreuse des os longs concerne l’architecture que l’on peut observer lorsque l’échantillon concerne un fragment de tissu prélevé de la périphérie vers le centre de la lésion : les travées osseuses se révèlent minces, éparpillées et réticulaires au centre de la lésion, augmentant en nombre et en taille à mesure que l’on se rapproche de la périphérie, pour devenir lamellaires et s’anastomoser entre elles jusqu’à ce qu’elles finissent par se confondre avec l’os hôte les entourant.
L’architecture organisée, typique des lésions réactives telles que les myosites ossifiantes, semble suggérer une nature réactive et réparatrice de la dysplasie ostéofibreuse des os longs.
On observe parfois une zone de cartilage hyalin, qui est probablement due à un cal réparateur d’une fracture de fatigue.
Immunohistochimie :
Les études immunohistochimiques avec anticorps de type cytokératines se sont révélées positives dans près de 85 % des cas.
Des cellules isolées et éparpillées et de petits tubes épithéliaux ont été observés, évoquant la relation possible de cette lésion avec l’adamantinome.
Cependant, les cellules épithéliales sont habituellement très éparpillées et difficiles à identifier à l’aide d’un colorant de type hématoxyline-éosine.
Campanacci et Laus n’ont retrouvé aucune cellule épithéliale dans les prélèvements biopsiques de leur série de 35 cas avec les colorants hématoxyline-éosine ; les mêmes observations ont été confirmées par d’autres études.
À l’inverse, aucune coloration positive à la cytokératine n’a jamais été retrouvée dans d’autres lésions ostéofibreuses telles que la dysplasie fibreuse et le fibrome ossifiant crânien, ce qui confirme l’histogenèse spécifique de la dysplasie ostéofibreuse des os longs et ses relations avec l’adamantinome.
Ces deux affections montrent une réaction positive à la vimentine dans les cellules fusiformes et les ostéoblastes du stroma fibreux.
Dans la dysplasie ostéofibreuse des os longs, le constituant épithélial s’est révélé positif aux cytokératines CKAE1/AE3 et CK19 et négatif pour les CK8 et CK18, tandis que dans l’adamatinome, une réaction positive inconstante a été observée, même aux deux dernières cytokératines.
Dans une autre étude, la dysplasie ostéofibreuse des os longs s’est révélée négative à l’antigène épithélial membranaire, alors que l’adamantinome a montré une réaction positive constante.
Analyse cytogénétique :
Des recherches récentes ont, à l’aide de l’analyse cytogénétique, confirmé la relation entre dysplasie ostéofibreuse des os longs et adamantinome.
En analysant des cultures, à court terme, de dysplasie ostéofibreuse des os longs au moyen de procédures cytogénétiques standards, et en utilisant une hybridation in situ fluorescente sur des cellules non cultivées, une trisomie des chromosomes 7, 8, 12 et 22 a été observée pour la dysplasie ostéofibreuse des os longs, suggérant une origine clonale et vraisemblablement néoplasique de cette lésion.
À la même époque, une trisomie des chromosomes 7, 8 et 12 a été décrite pour l’adamantinome, ce qui tend à prouver que les deux lésions ont une histogenèse commune.
Pathogenèse :
Ces 10 dernières années, la pathogenèse de la dysplasie ostéofibreuse des os longs et ses relations avec l’adamantinome ont fait l’objet de nombreuses discussions.
Les colorations immunohistochimiques à l’aide d’anticorps de cytokératines ont révélé la présence d’un composant épithélial modeste dans la plupart des cas de dysplasie ostéofibreuse des os longs.
Par ailleurs, dans l’adamantinome classique, certaines zones présentant l’aspect histologique typique de la dysplasie ostéofibreuse des os longs sont couramment observées à la périphérie de la lésion, associées à un tissu épithélial malin.
Dans ces deux lésions, le tissu ostéofibreux semble être doté d’un pouvoir réparateur, et l’on pense que sa prolifération pourrait être une réaction au composant épithélial.
Ces observations permettent de présumer que la dysplasie ostéofibreuse des os longs et l’adamantinome partagent une même origine histogénétique.
En 1989, Czerniak et al ont émis l’hypothèse selon laquelle la dysplasie ostéofibreuse pourrait être le résultat d’un processus réparateur secondaire dont la croissance surpasserait celle du tissu tumoral épithélial arrivé à maturité et en régression (en se différenciant), nommant cette lésion « adamantinome différencié ».
Selon Mirra, qui a défini la dysplasie ostéofibreuse des os longs comme un « adamantinome juvénile », les îlots de cellules épithéliales contenus dans la dysplasie ostéofibreuse des os longs pourraient, après de nombreuses années, être à l’origine d’une tumeur maligne.
L’absence de réponse positive des cellules épithéliales aux cytokératines a été observée dans quelques 15 % des cas et pourrait être due à la défaillance de l’analyse immunohistochimique provoquée par la décalcification du tissu ou par l’exiguïté du nombre de cellules contenant des filaments de kératine.
Par ailleurs, on a avancé qu’il pourrait exister une forme de dysplasie ostéofibreuse des os longs sans relation avec l’adamantinome.
Une autre hypothèse est que les cellules épithéliales pourraient parfois régresser spontanément, en étant « débordées » par le tissu ostéofibreux réactif.
Pour résumer ces théories récentes, la dysplasie ostéofibreuse des os longs semble être un hamartome provenant d’un reliquat épithélial ectodermique « délocalisé » dans la corticale au cours du développement embryonnaire.
Le tissu réparateur ostéofibreux semble réagir à ces éléments épithéliaux en maintenant une sorte d’équilibre au cours de l’enfance (de 0 à 10 ans) : il peut se produire une progression clinique et radiographique (« adamantinome différencié », positif aux cytokératines) ou une régression de la lésion (« dysplasie ostéofibreuse », négative aux cytokératines).
In fine, la lésion tendrait vers un processus autorésolutif après la puberté.
Mais parfois, au cours de l’enfance, le composant épithélial peut progresser et devenir histologiquement détectable grâce à une coloration à l’hématoxyline et à l’éosine.
Les cellules épithéliales forment un petit îlot et se mêlent au stroma classique de la dysplasie ostéofibreuse des os longs.
Cette lésion a été dénommée « adamantinome intracortical juvénile » ou « adamantinome semblable à la dysplasie ostéofibreuse » (moins de 20 cas ont été rapportés dans la littérature).
L’évolution d’une dysplasie ostéofibreuse des os longs avec cellules épithéliales positives aux cytokératines (adamantinome régressif) vers un adamantinome classique après récidive a été rapportée dans deux cas.
À l’exception de ces deux cas, l’évolution d’une dysplasie ostéofibreuse des os longs vers un adamantinome classique n’a jamais été observée dans aucune des autres séries rapportées dans la littérature ayant bénéficié d’un suivi étendu.
Diagnostic différentiel :
Le diagnostic différentiel avec la dysplasie fibreuse est généralement facile.
La dysplasie ostéofibreuse des os longs n’est jamais polyostosique (on n’a décrit qu’un seul cas multicentrique et d’ailleurs sans confirmation histologique), hormis le fait qu’elle peut concerner à la fois tibia et péroné ou être bilatérale.
Tibia et péroné sont des sites inhabituels pour la dysplasie fibreuse, qui est plus fréquente au fémur.
Sur les radiographies, la dysplasie fibreuse est faite d’une ostéolyse intramédullaire centrale délimitée de façon inconstante par un liseré d’ostéosclérose, contenant une opacité en « verre dépoli » typique.
La déformation tibiale, caractéristique de la dysplasie ostéofibreuse des os longs, ne se rencontre jamais dans la dysplasie fibreuse.
D’un point de vue histologique, la dysplasie fibreuse présente un tissu fibreux plus cellulaire, contenant des travées réticulaires non bordées d’ostéoblastes actifs.
Il n’existe pas d’architecture organisée, les travées osseuses restent réticulaires, même à la périphérie de la lésion, et ne se transforment pas progressivement en os lamellaire, trait typique de la dysplasie ostéofibreuse des os longs.
Le diagnostic différentiel avec l’adamantinome du tibia est un problème bien plus réel, dans la mesure où ce dernier peut présenter le même aspect radiographique que la dysplasie ostéofibreuse des os longs.
Parfois, un prélèvement biopsique sur les zones périphériques de l’adamantinome peut montrer un aspect histologique ressemblant étroitement à la dysplasie ostéofibreuse des os longs, sans toutefois contenir l’élément épithélioïde déterminant pour le diagnostic.
Dans ces cas, le diagnostic différentiel est fondé sur les notions cliniques et radiographiques : une lésion progressant radiographiquement après la puberté et devenant douloureuse fait soupçonner un adamantinome.
En outre, la dysplasie ostéofibreuse des os longs est habituellement une lésion intracorticale, alors que l’adamantinome comporte volontiers une extension intramédullaire et un envahissement des parties molles.
Dans de telles situations, une biopsie est impérative.
Plus couramment, à l’histologie, l’adamantinome comporte des amas épithéliaux hyperchromatiques évidents tandis que, dans la dysplasie ostéofibreuse des os longs, l’immunocoloration par cytokératine ne présente que des îlots isolés de cellules épithéliales.
Selon Czerniak et al, il existe une forme intermédiaire entre la dysplasie ostéofibreuse des os longs et l’adamantinome malin classique, qu’ils ont nommée « adamantinome différencié », laquelle se caractérise par l’âge du patient (20 premières années), la localisation intracorticale et la prédominance histologique d’un aspect semblable à la dysplasie ostéofibreuse des os longs typique avec des éléments épithéliaux éparpillés positifs à la cytokératine.
Les auteurs ont montré qu’il pouvait y avoir deux évolutions différentes possibles de cette lésion intermédiaire :
– le tissu ostéofibreux réactif pourrait croître davantage que le tissu tumoral à maturité et en régression, évoluant vers une résolution spontanée ;
– le tissu épithélial tumoral prend le dessus, évoluant vers un adamantinome classique.
Jusqu’à présent, l’évolution d’un adamantinome différencié ou régressif vers un adamantinome malin au sein d’une récidive locale n’a été rapportée que deux fois dans la littérature.
Évolution et traitement :
Le comportement évolutif de la dysplasie ostéofibreuse des os longs est variable et imprévisible, pouvant aller d’une petite lésion latente à une ostéolyse proliférative progressive conduisant in fine à des déformations du squelette.
En général, la lésion a une croissance lente et modérée au cours des 5 premières années de la vie. Une diminution de la vitesse de prolifération est généralement observée entre 5 et 10 ans, avec stabilisation de l’aspect radiologique et des déformations.
Enfin, après l’âge de 10-12 ans, l’ostéolyse tend à s’ossifier progressivement, conduisant à une guérison spontanée en fin de puberté.
Cette guérison spontanée peut parfois intervenir plus précocement au cours de l’enfance.
Généralement, à l’âge adulte, le seul reliquat d’une dysplasie ostéofibreuse des os longs non traitée est un tibia incurvé ressemblant à un tibia pagétique.
Par le passé, la dysplasie ostéofibreuse des os longs a été considérée comme une maladie agressive du fait de sa tendance à la récidive après curetage ou résection.
En effet, lorsqu’une intervention chirurgicale a lieu avant l’âge de 10 ans, les récidives locales sont très communes, et parfois une nouvelle lésion dysplasique apparaît en un autre point du même os.
Le taux de récidive locale mentionné dans la littérature est compris entre 16 et 64 %.
En 1981, Campanacci et Laus ont rapporté 16 récidives locales sur 25 patients traités par curetage ou résection sous-périostée, avec ou sans greffe osseuse.
Aucune de ces récidives n’a été observée après l’âge de 10 ans, et l’évolution des lésions récidivantes s’est faite sous la forme d’une croissance lente aboutissant à une stabilisation complète à la puberté.
Habituellement, aucun traitement chirurgical n’est nécessaire.
Lorsque la lésion est indolore et qu’elle présente l’aspect radiographique typique d’une dysplasie ostéofibreuse des os longs chez l’enfant de moins de 10 ans, la biopsie elle-même n’est pas toujours nécessaire.
Cependant, des adamantinomes classiques et semblables à la dysplasie ostéofibreuse ont été décrits chez des enfants de moins de 10 ans.
L’enfant doit donc être suivi tout au long de sa croissance et une biopsie doit être envisagée en cas de progression radiographique agressive (invasion intramédullaire, extension vers les tissus mous) ou lorsque des douleurs apparaissent.
Le traitement chirurgical doit être réservé aux lésions qui progressent radiographiquement de façon nette, aux patients présentant des déformations squelettiques ou un risque de fracture pathologique.
Lorsque le risque de fracture est très élevé, il est possible de recourir à une protection par orthèse externe, mais un traitement chirurgical avec curetage et greffe osseuse est généralement préférable.
Lorsque les déformations squelettiques sont sévères, une ostéotomie de correction peut être réalisée, de préférence après la puberté.







")



{kind=link}