



















Introduction : poches pharyngobranchiales ou poches endobranchiales
Chaque arc branchial du foetus est constitué d’un axe mésenchymateux tapissé extérieurement d’ectoblaste et intérieurement d’entoblaste.
En plus du mésenchyme d’origine locale, l’axe des arcs reçoit un contingent cellulaire migrant à partir de la crête neurale.
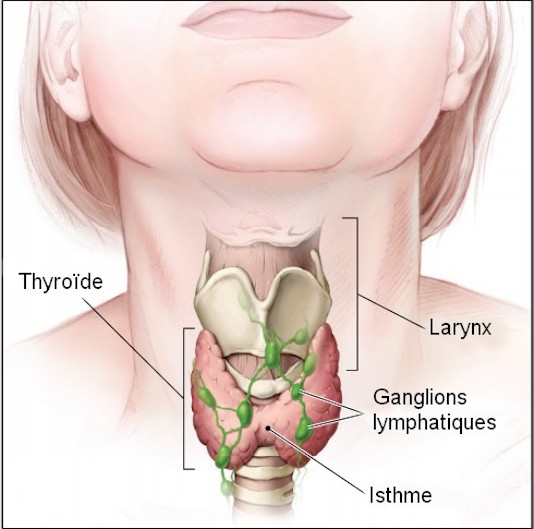 Du fait de la présence des arcs branchiaux, on peut distinguer chez l’embryon humain cinq paires de poches endobranchiales.
Du fait de la présence des arcs branchiaux, on peut distinguer chez l’embryon humain cinq paires de poches endobranchiales.
La dernière de ces poches est atypique et souvent considérée comme un diverticule de la quatrième.
Le revêtement endoblastique de ces poches donne naissance à un certain nombre d’organes importants.
Nous étudierons ici le devenir des troisième, quatrième et cinquième poches, qui donnent naissance aux parathyroïdes et participent à l’élaboration de la glande thyroïde.
Ontogenèse des hormones thyroïdiennes :
Ces dernières années ont permis de mieux comprendre l’ontogénie du système thyroïdien chez le foetus.
De récentes avancées en biologie du développement ont ainsi permis d’analyser ses mécanismes moléculaires.
Cet article comprend une synthèse des connaissances actuelles sur le développement du système thyroïdien foetal, centrée sur le foetus humain.
A – DÉVELOPPEMENT DU SYSTÈME THYROÏDIEN FOETAL :
La maturation de l’axe hypothalamo-hypophysaire et thyroïdien du foetus humain commence pendant le premier trimestre de gestation et se poursuit au cours des premiers mois de la vie postnatale.
Les premiers noyaux hypothalamiques et ceux de la zone supraoptique sont visibles à 12 semaines de développement, tandis que le reste des noyaux hypothalamiques et de l’éminence médiane sont présents dès 16 semaines de développement.
La thyrotropin releasing hormone (TRH) est présente dans des extraits hypothalamiques à 10 semaines de développement.
Les capillaires caractéristiques du système porte hypothalamo-hypophysaire sont détectés dès 16 semaines de développement.
La maturation anatomique du système porte hypothalamohypophysaire est complète à 30-35 semaines de développement.
Des granules de sécrétion peuvent être identifiés dans les cellules de l’antéhypophyse à 10-12 semaines de développement, date à laquelle l’hypophyse est déjà partiellement située dans une cavité osseuse, la selle turcique.
En utilisant des bio-essais et des radio-immuno-essais, la thyrotropine (thyroxine stimulating hormone [TSH]) peut être détectée dès 10-12 semaines de développement.
La glande thyroïde se développe à partir, d’une part d’une évagination ventrale du plancher buccopharyngé sur la ligne médiane (ébauche médiane), et d’autre part des extensions caudales et bilatérales des quatrièmes poches pharyngobranchiales (ébauches latérales, parfois appelées cinquièmes poches ou corps ultimobranchiaux).
Les cellules des corps ultimobranchiaux donnent naissance aux cellules parafolliculaires ou cellules C de la glande thyroïde, qui sécrètent la calcitonine.
Ces structures sont visibles dès 16-17 jours de développement.
À 7 semaines de développement, l’ébauche médiane et les ébauches latérales ont fusionné et la thyroïde a migré dans sa localisation définitive à la partie antérieure du cou.
À 10 semaines de développement, l’accumulation de colloïde peut déjà être détectée dans les cellules folliculaires thyroïdiennes et la synthèse de thyroglobuline a commencé.
La maturation structurale de la glande thyroïde est pratiquement achevée à 17 semaines de développement, date à laquelle la glande thyroïde pèse environ 300 mg.
Certains facteurs de transcription jouant probablement un rôle important dans la mise en place de la glande thyroïde ont été récemment identifiés.
Il s’agit du thyroid transcription factor (TTF)1, TTF2 et Pax-8.
Des travaux récents réalisés in vitro mais aussi in vivo, notamment chez la souris, étudiant l’invalidation de ces gènes, ont permis de mieux comprendre leur fonction.
TTF1 et Pax-8 semblent impliqués dans la différenciation fonctionnelle terminale de la glande thyroïde en se fixant aux domaines de contrôle des gènes codant pour la thyroglobuline et la thyroperoxydase.
TTF2 semble agir en amont de TTF1 et de Pax-8 et pourrait être un répresseur de l’expression de TTF1 et de Pax-8 empêchant donc la différenciation fonctionnelle terminale des cellules thyroïdiennes tant que la thyroïde n’a pas migré en bonne place ; l’extinction de son expression permettant alors l’expression des marqueurs de différenciation fonctionnelle terminale.
L’invalidation génique de TTF1, TTF2 ou Pax-8 entraîne chez la souris des anomalies du développement de type athyréose, ectopie ou hypoplasie thyroïdienne.
Chez l’homme, certains de ces gènes ont aussi été retrouvés chez des patients atteints d’hypothyroïdie congénitale.
Ainsi, ces facteurs de transcription participent au développement de la thyroïde mais leur rôle exact et leur responsabilité dans les dysgénésies thyroïdiennes restent à préciser.
Par ailleurs, le facteur de transcription TTF1 semble aussi impliqué dans le développement des cellules thyréotropes, suggérant un développement combiné entre l’organecible (la thyroïde) et l’organe de commande (l’hypophyse).
Les gènes Hox du groupe 3 semblent aussi jouer un rôle dans la détermination de la glande thyroïde.
La TSH ne semble pas jouer un rôle dans la migration de la thyroïde.
Elle joue cependant un rôle sur la croissance de la glande, une fois celle-ci en place dans la loge thyroïdienne.
Ceci est suggéré par l’observation de nouveau-nés anencéphales qui ont une glande thyroïde en situation normale mais hypoplasique.
De plus, des mutations inactivatrices du gène du récepteur de la TSH, responsables de résistances à la TSH ou d’hypothyroïdies congénitales, chez des patients porteurs d’une glande en place mais hypoplasique, ont été rapportées.
B – MATURATION DU SYSTÈME HYPOTHALAMOHYPOPHYSAIRE ET THYROÏDIEN FOETAL :
Les différentes étapes du développement du système hypothalamique, hypophysaire et thyroïdien permettant la maturation de la fonction thyroïdienne foetale, sont complexes et interdépendantes.
La glande thyroïdienne foetale est capable de concentrer l’iode et de synthétiser la iodothyronine à partir de 70 jours de développement (10 semaines de développement).
Toutefois, la production d’hormones thyroïdiennes par la thyroïde foetale reste limitée jusqu’à 18 à 20 semaines de développement.
À ce moment, la captation d’iode des cellules folliculaires thyroïdiennes augmente et la concentration sérique foetale de T4 commence à s’accroître.
La concentration de T4 totale et la concentration de T4 libre s’accroissent alors progressivement jusqu’aux dernières semaines de grossesse.
La concentration sérique de T3 reste basse jusqu’à 30 semaines de développement et s’accroît ensuite en deux phases, une phase prénatale et une phase postnatale.
L’accroissement prénatal de la concentration foetale de T3 est déterminée par la maturation de la désiodase de type 1 hépatique et par la conversion hépatique accrue de T4 en T3.
Cependant, d’autres lieux de conversion concourent aussi à cette augmentation, tels que la graisse brune et le rein.
La production de TRH hypothalamique mature entre 18 et 20 semaines de développement et le système vasculaire porte hypothalamohypophysaire entre 35 et 40 semaines de développement.
La sécrétion foetale de TSH au cours de la gestation peut atteindre des valeurs allant jusqu’à 10 mUI/mL à terme.
Chez le mouton, la réponse de la TSH à la stimulation par le TRH s’accroît progressivement pendant la deuxième partie de la gestation pour être à terme maximale.
La fonction thyroïdienne foetale se développe donc sous l’influence d’une concentration de TSH qui s’accroît dans la deuxième partie de la gestation.
L’accroissement du ratio T4/TSH, associé avec une augmentation progressive de la concentration de T4 libre au cours du dernier trimestre, suggère des modifications, d’une part dans la sensibilité des cellules thyréotropes hypophysaires au rétrocontrôle négatif des hormones thyroïdiennes, et d’autre part dans la sensibilité des cellules folliculaires thyroïdiennes à la TSH.
Ainsi, il semble exister une maturation progressive de la réponse thyroïdienne à la TSH chez le foetus.
Celle-ci a pu être documentée chez le mouton et semble probable chez le foetus humain. Le processus de synthèse des hormones thyroïdiennes est donc plus ou moins immature jusqu’à 30 à 35 semaines de développement.
Les concentrations de thyroglobuline par unité de poids du tissu thyroïdien sont faibles, tandis que la concentration d’iode inorganique est assez élevée, ce qui suggère l’immaturité du processus d’organification.
À terme, la glande thyroïdienne est fonctionnellement mature, pèse 1 à 1,5 g en l’absence de carence iodée et le volume thyroïdien est d’environ 1 mL.
Le contrôle de la sécrétion des hormones thyroïdiennes foetales peut donc être caractérisé comme une balance entre trois facteurs qui augmentent au cours de la gestation : la sécrétion de TRH hypothalamique, la sensibilité des cellules folliculaires thyroïdiennes à la TSH et la sensibilité des cellules hypophysaires à l’inhibition des hormones thyroïdiennes sur la sécrétion de TSH.
D’un point de vue fonctionnel, le foetus est en situation d’hypothyroïdie primaire et tertiaire (à la fois d’origine thyroïdienne et hypothalamique) au milieu de la gestation.
Il évolue ensuite vers un stade d’hypothyroïdie tertiaire modérée pendant les dernières semaines de gestation et enfin vers une fonction thyroïdienne mature au cours de la période périnatale.
Après la naissance, la concentration de T3 et de T4 augmente de deux à six fois au cours des premières heures de vie avec un pic 24 à 36 heures après la naissance.
Ces concentrations décroissent ensuite jusqu’au niveau caractéristique des nourrissons dans les 4 à 5 premières semaines de vie.
Dans les premières minutes de la vie postnatale, la TSH augmente brutalement, avec un pic sérique atteint à 30 minutes de vie. La concentration de la TSH circulante reste ensuite modérément élevée pendant 2 à 3 jours.
L’augmentation de sécrétion de la T4 sérique, observée immédiatement après la naissance, est dépendante de ce pic de TSH et est due à une sécrétion accrue par la thyroïde du nouveau-né.
En ce qui concerne l’accroissement de la concentration de T3 dans des zones d’hyperthyroïdie pour un adulte, elle est d’une part due à la stimulation par la TSH de la production de T3 thyroïdienne, et d’autre part à la maturation rapide de la désiodase de type 1, ainsi qu’à la conversion de T4 en T3 en période néonatale.
C – MATURATION DU MÉTABOLISME DES HORMONES THYROÏDIENNES FOETALES :
La déiodination des iodothyronines est la principale voie métabolique.
La désiodase de type 2 convertit la T4 en T3 et la reverse T3 en T2, et la désiodase de type 3 modifie la T4 en reverse T3 et la T3 en T2.
Ainsi, chez l’adulte, 70 à 90 % de la T3 circulante vient de la conversion périphérique de T4 et 10 à 30 % vient de la sécrétion thyroïdienne.
La reverse T3 vient quasiment en totalité de la conversion périphérique de T4.
La T3 et la reverse T3 sont progressivement déiodinées en composés dépourvus d’activité biologique.
Les désiodases de types 2 et 3 sont présentes dans les tissus foetaux dès le milieu de la gestation tandis que la désiodase de type 1 n’est pas encore exprimée.
En conséquence, pendant le troisième trimestre, la concentration sérique foetale de T3 est basse, et les concentrations de reverse T3, de T4 sulfate, de T3 sulfate et de reverse T3 sulfate sont hautes.
La reverse T3, la T4 sulfate et la reverse T3 sulfate sont biologiquement inactives.
Le sulfate de T3 est aussi inactif, mais pourrait servir de source locale de T3 dans les tissus foetaux contenant une activité sulfatase.
La sulfoconjugaison est donc une voie métabolique possible pour les iodothyronines.
Chez le foetus, il est maintenant clair que cette voie métabolique est majeure.
Chez les foetus de rats et de moutons, pendant la deuxième partie de la gestation, le cerveau, le tissu adipeux brun, l’hypophyse et peut-être la peau, contiennent des niveaux significatifs de monodésiodase de type 2 qui convertit localement la T4 en T3.
Chez le foetus hypothyroïdien, l’activité de la désiodase de type 2 est accrue dans ces tissus tandis que les désiodases de types 1 à 3 ont des activités diminuées.
Ainsi, le métabolisme des hormones thyroïdiennes, contrôlé par l’expression et l’activité des enzymes désiodases, est caractérisé par la production prédominante d’analogues inactifs.
Cette inactivation permet probablement de minimiser le catabolisme foetal et d’augmenter l’anabolisme dans la plupart des tissus foetaux.
Chez les foetus hypothyroïdiens, l’activité des désiodases est modifiée de manière à permettre l’accès de la T4 aux tissus cérébraux dans lesquels la déiodination en T3 est accrue et la dégradation de la T3 diminuée.
Dans cette situation d’hypothyroïdie, le transfert maternofoetal limité de T4 permet de maintenir des concentrations cérébrales de T3 satisfaisantes et pourrait ainsi protéger la maturation cérébrale du foetus hypothyroïdien.
D – INTERACTIONS MÈRE-FOETUS :
Des données indirectes sont venues de l’analyse des nouveau-nés après que le dépistage systématique de l’hypothyroïdie congénitale ait été mis en place.
Il apparaît que la taille, le poids, l’aspect, le comportement, l’adaptation à la vie extra-utérine et le développement postnatal immédiat sont normaux chez des nouveau-nés hypothyroïdiens, même en cas d’agénésie thyroïdienne.
Néanmoins, la maturation osseuse est retardée à la naissance chez environ 70 % des nouveau-nés ayant une hypothyroïdie congénitale.
De plus, certains nouveau-nés peuvent avoir des signes cliniques discrets d’hypothyroïdie, mais ceux-ci ne permettent pas en règle de faire le diagnostic dans la période néonatale.
En effet, les signes classiques d’hypothyroïdie se développent progressivement, en l’absence de traitement, pendant les premières semaines ou mois de la vie extra-utérine.
Ces observations démontrent que malgré un transfert maternofoetal d’hormones thyroïdiennes en cas d’hypothyroïdie congénitale, il existe un certain degré d’hypothyroïdie foetale.
Un cas rapporté démontre bien qu’en l’absence de transfert maternofoetal de T4, l’hypothyroïdie congénitale entraîne des anomalies de maturation cérébrale, même en cas de traitement postnatal rapide.
Il s’agissait d’une mutation d’un facteur de transcription, Pit.1 (qui gouverne entre autre l’expression de la TSH), présente chez la mère et son foetus qui entraînait une hypothyroïdie maternelle et foetale.
Un retard du développement intellectuel était constaté chez le nouveau-né malgré un traitement postnatal immédiat par hormones thyroïdiennes.
À l’inverse, même en l’absence d’hypothyroïdie congénitale et avec une fonction thyroïdienne foetale normale, il semble que le transfert maternofoetal de T4 joue un rôle dans le développement cérébral du foetus.
En effet, le développement psychomoteur des enfants issus de mères ayant un certain degré d’hypothyroïdie pendant la grossesse est moins bon que celui des enfants issus de mères euthyroïdiennes pendant leur grossesse.
Ceci est un argument de plus qui souligne que même si la fonction thyroïdienne foetale est relativement indépendante de la fonction thyroïdienne maternelle, il existe des échanges, en particulier de T4, entre le foetus et la mère.
E – RÔLE DU PLACENTA DANS LE DÉVELOPPEMENT ET LA FONCTION DE LA THYROÏDE FOETALE :
Le placenta humain est une barrière qui permet peu le transfert maternofoetal d’hormones thyroïdiennes ; aussi la maturation du système thyroïdien foetal s’effectue de manière relativement indépendante de l’influence maternelle.
Le placenta humain est imperméable à la thyréotropine mais est perméable à l’iode.
L’iode est une substance essentielle pour la synthèse des hormones thyroïdiennes.
En conséquence, un apport adéquat d’iode maternel est essentiel pour la production foetale d’hormones thyroïdiennes.
Celui-ci est important en particulier pendant la deuxième partie de la gestation moment durant lequel la production d’hormones thyroïdiennes foetales augmente progressivement.
Pendant le premier trimestre, la cavité amniotique, qui contient l’embryon, est entourée du coelome extraembryonnaire qui contient le liquide coelomique.
Le coelome extraembryonnaire est entouré du placenta.
Avant que le système hypothalamohypophysaire et thyroïdien foetal ne devienne fonctionnel, entre 6 et 12 semaines de développement, il existe un gradient de concentration des hormones thyroïdiennes qui est maximal dans le sérum maternel, intermédiaire dans le liquide coelomique, et faible dans le liquide amniotique.
Ceci montre que avant que la vascularisation placentaire ne soit pleinement fonctionnelle, les hormones thyroïdiennes maternelles ont accès à l’embryon.
Pendant les deuxième et troisième trimestres de gestation, il existe un gradient maternofoetal des hormones thyroïdiennes important avec une concentration de T4 libre et de T3 libre bien plus importante chez la mère.
Ce gradient diminue progressivement au fur et à mesure que la fonction thyroïdienne foetale mature et que l’on se rapproche du terme de la grossesse.
Cependant, à la naissance, il persiste et l’on observe une concentration de T3 libre deux à trois fois plus élevée dans le sérum de la mère que dans le sang du cordon.
Par ailleurs à terme, chez les nouveau-nés ayant une athyréose ou une absence complète de production d’hormones thyroïdiennes, comme cela est décrit dans les anomalies génétiques de synthèse de la thyroglobuline, le taux de T4 libre au sang du cordon n’est pas nul et représente approximativement 30 % du taux de T4 présent au stade foetal.
Ces données indiquent la présence d’un transfert significatif, bien que relativement limité, d’hormones thyroïdiennes maternelles vers le compartiment foetal tout au long de la grossesse.
Il n’y a cependant pas de données quantifiant exactement ce transfert de T4 libre qui est probablement très variable d’un sujet à l’autre.
Le TRH est transporté à travers le placenta du compartiment maternel vers le compartiment foetal, mais le taux circulant maternel très bas du TRH ne permet pas un transfert quantitativement significatif.
Le TRH n’est pas exclusivement synthétisé par l’hypothalamus mais aussi par le placenta, par le pancréas foetal et peut-être par d’autres tissus du tractus digestif.
Ainsi, les concentrations foetales plasmatiques et tissulaires de TRH sont relativement élevées en particulier pendant le premier et le deuxième trimestre de gestation, du fait de cette production extrahypothalamique.
L’activité faible de dégradation du TRH dans le sérum foetal contribue aussi au taux élevé de TRH chez le foetus. Hors la production hypothalamique du TRH mature tardivement, à proximité du terme.
La présence de TRH à haute concentration chez des foetus de moutons, la modulation du taux sanguin, pancréatique et placentaire par les hormones thyroïdiennes chez ces animaux suggèrent un rôle du TRH extrahypothalamique dans le contrôle de la sécrétion de la TSH foetale.
Le rôle du TRH extrahypothalamique chez le foetus humain reste à préciser.
Le placenta produit de larges quantités d’human chorionic gonadotropin (hCG).
Cette hCG a probablement pour rôle principal la maintenance du corps jaune pendant la première partie de la gestation, la stimulation des testicules chez le foetus et la stimulation de la production de progestérone placentaire.
L’hCG a également une activité biologique similaire à la TSH mais cette activité est faible, même si elle joue probablement un rôle dans l’adaptation de la thyroïde maternelle au cours de la grossesse.
Cependant, l’hCG sécrétée dans la circulation foetale est en faible concentration et son rôle dans la maturation et la fonction du système thyroïdien foetal est probablement faible.
Le placenta est le siège de synthèse d’enzymes à activité désiodase qui catalyse la déiodination de la T4 et de la T3.
La désiodase de type 2 et la désiodase de type 3 sont exprimées fortement dans le placenta.
Ainsi on observe dans le tissu placentaire la conversion de T4 en T3, de T4 en reverse T3 et de T3 en T2.
Ceci permet l’inactivation par le placenta de la plupart de la T4 et de la T3 arrivant par la face maternelle ou foetale de la circulation sanguine.
De plus, cette activité des désiodases placentaires est une source secondaire et continue d’iode pour le foetus.
En situation d’hypothyroïdie l’activité de la désiodase de type 2 s’accroît notamment dans le placenta, permettant l’augmentation de la production de T3.
La plupart de la T3 produite par la conversion placentaire de la T4 par le placenta est probablement active seulement localement à cause de la présence de l’activité de la désiodase de type 3.
En conséquence, il semble que le placenta fonctionne d’une part pour maintenir un taux bas de T3 chez le foetus, et d’autre part pour maintenir un apport constant de la forme active de la T3 aux cellules déciduales.
F – CONCLUSION :
L’axe hypothalamohypophysaire et thyroïdien foetal se développe relativement indépendamment de la fonction thyroïdienne maternelle.
Il est néanmoins dépendant des échanges placentaires en particulier pour un apport d’iode adéquat et un transfert maternofoetal de thyroxine, significatif tout au long de la gestation.
Cette contribution maternelle à la thyroxine foetale est importante pour la maturation normale du foetus et particulièrement de son système nerveux central.
Ontogenèse des hormones parathyroïdiennes :
A – DÉVELOPPEMENT DES PARATHYROÏDES :
Les troisième et quatrième poches endobranchiales présentent à leur extrémité distale deux recessus, un ventral et un dorsal.
À la cinquième semaine de développement, l’épithélium du recessus dorsal de la troisième poche se différencie en tissu parathyroïdien, tandis que le recessus ventral forme l’ébauche du thymus.
Les ébauches glandulaires perdent leurs connexions avec la paroi pharyngienne et la demi-ébauche thymus migre en direction caudale et médiale, entraînant avec lui la parathyroïde.
Le corps thymique rejoint rapidement sa situation définitive dans le thorax, où il fusionne avec sa demi-ébauche homologue du côté opposé.
Sa portion caudale, étroite et allongée, se segmente en petits fragments qui disparaissent habituellement.
Le tissu parathyroïdien de la troisième poche endobranchiale vient en définitive reposer sur la face dorsale du corps thyroïde, pour former chez l’adulte la glande parathyroïde inférieure.
Le revêtement épithélial du recessus dorsal de cette poche donne la glande parathyroïde supérieure.
Après avoir perdu ses connexions avec la paroi du pharynx, la parathyroïde supérieure vient s’amarrer au corps thyroïde qui effectue sa migration en direction caudale.
Elle se retrouve ainsi située à la face dorsale de la glande thyroïde.
Il peut exister des anomalies de migration des parathyroïdes supérieures et inférieures.
Les parathyroïdes supérieures peuvent migrer dans la partie inférieure de la glande thyroïde ; les parathyroïdes inférieures peuvent, quant à elles, migrer de la fourchette sternale à l’intérieur du thymus, ou encore dans la glande thyroïde.
Les gènes impliqués dans le développement et la migration des parathyroïdes sont inconnus.
Les études sur des animaux transgéniques ont permis de montrer que le gène homéotique Hox 1.5 pouvait être impliqué dans le développement des parathyroïdes.
En effet, l’invalidation du gène Hox 1.5 chez la souris entraîne une anomalie de développement des troisième et quatrième arcs branchiaux responsable d’un phénotype similaire à celui existant dans le syndrome de Di George, associant à une hypoplasie parathyroïdienne, des anomalies cardiaques et une aplasie thymique.
D’autres gènes Hox jouent un rôle dans le développement du thymus, de la thyroïde et des parathyroïdes.
L’invalidation du gène Hox a3 chez la souris entraîne des anomalies thyroïdiennes telles que des formes inhabituelles de l’isthme thyroïdien ou des hémiagénésies.
Cependant, elle n’est jamais responsable d’agénésie.
L’addition à ces souris mutantes d’invalidation des gènes paralogues de Hox a3, Hox b3 et/ou d3, exacerbe le phénotype mais toujours sans jamais donner d’agénésie thyroïdienne.
Il est probable que ces gènes agissent surtout dans les corps ultimobranchiaux puisque c’est leur lieu d’expression.
De plus, chez les souris invalidées, pour les gènes Hox b3 et Hox d3, la présence à l’état hétérozygote du gène Hox a3 entraîne des anomalies de migration du thymus et des glandes parathyroïdes.
Très récemment, le rôle d’un gène codant pour un facteur de transcription, glial cells missing 2 (Gcm2), a été démontré dans l’ontogénie des glandes parathyroïdes.
En effet, l’invalidation génique de ce gène entraîne l’absence de développement des glandes parathyroïdes.
Ces souris sont viables bien qu’elles aient une hypoparathyroïdie.
En effet, chez ces souris, le thymus est une source de suppléance partielle de parathormone.
B – PHYSIOLOGIE DES PARATHYROÏDES FOETALES : SYSTÈME CALCITONINE, PARATHORMONE
Les glandes parathyroïdes et les cellules C de la thyroïde sont identifiables dès la fin du premier trimestre de gestation. Ces deux systèmes sont fonctionnels durant les deuxième et troisième trimestres de grossesse.
Les études faites chez le mouton, le singe et le foetus humain montrent que des concentrations élevées de calcium (2,75 à 3 mmol/L au dernier trimestre) sont maintenues par un transfert placentaire actif de la mère vers le foetus.
Les concentrations de PTH du sang de cordon au troisième trimestre sont relativement basses tandis que celles de calcitonine sont élevées. Ni la calcitonine, ni la PTH ne sont transportées au travers du placenta.
Le 25-hydroxycholécalciférol et le 1-25-hydroxycholécalciférol sont transportés au travers du placenta et leurs concentrations sont égales ou supérieures à celles du sang maternel.
Les taux élevés de calcium total et ionisé maintenus par transfert actif de la mère au foetus entraînent une diminution du taux de PTH et augmente la sécrétion de calcitonine par les cellules C de la thyroïde. Les glandes parathyroïdes contiennent aussi des taux élevés de PTHrp.
Cette dernière hormone est présente dans le placenta et de nombreux tissus foetaux.
Elle joue un rôle majeur dans le développement osseux foetal et dans l’homéostasie calcique du foetus.
Elle se fixe aussi au récepteur de la PTH par la partie de la protéine contenant les 34 premiers acides aminés.
Par d’autres parties de la molécule, elle inhibe l’activité des ostéoclastes et stimule le transfert calcique au travers du placenta. La PTH et la PTHrp agissent sur le rein foetal pour augmenter la réabsorption du calcium.
Le sang de cordon humain contient des concentrations plus élevées de PTHrp que les échantillons maternels prélevés au même moment chez les mères.
Ceci plaide pour un rôle de la PTHrp dans le contrôle du métabolisme calcique foetal. La PTH et la PTHrp stimulent la production rénale foetale de 1-25- hydroxycholécalciférol, qui, à son tour accroît le transfert transplacentaire de calcium au bénéfice du foetus.
En conclusion, l’axe entre le placenta et la glande parathyroïde, qui est fonctionnel chez le foetus, favorise le transfert maternofoetal de calcium et l’accrétion osseuse chez le foetus. Les taux importants de calcitonine chez le foetus sont importants pour l’inhibition de la résorption osseuse foetale et stimule donc l’anabolisme osseux.
La PTHrp joue un rôle prédominant dans le contrôle du métabolisme phosphocalcique foetal.







")



{kind=link}