



















Historique :
En 1859, pour la première fois le diagnostic d’alcaptonurie a été évoqué par Boedeker chez un patient dont les urines noircissaient lorsqu’on leur ajoutait de « l’alcalie ».
C’est en raison de la propriété qu’avaient ces urines de fixer intensément l’oxygène en milieu alcalin que Boedeker créa le mot Alkapton auquel s’ajoutait quelque temps plus tard le terme d’Alkaptonurie (alcaptonurie en français).
 Boedeker ne put reconnaître la nature chimique de cette substance avide d’oxygène.
Boedeker ne put reconnaître la nature chimique de cette substance avide d’oxygène.
Ce n’est que plus de 30 ans plus tard que Wolkow et Baumann l’identifièrent et la nommèrent « acide homogentisique » en raison de sa structure chimique très voisine de celle de l’acide gentisique.
Compte tenu de la structure aromatique de l’acide homogentisique, on pensa rapidement qu’il provenait du métabolisme d’autres composés aromatiques et en particulier des acides aminés tels que la tyrosine et la phénylalanine entrant dans la composition des protéines alimentaires, ce qui fut vérifié expérimentalement.
En 1909, Neubauer proposa pour la première fois les maillons de la chaîne métabolique de la tyrosine qui, à quelques détails près, est demeurée inchangée jusqu’à nos jours.
En 1908, Garrod pensa que l’alcaptonurie était due à l’absence ou au défaut d’activité d’une enzyme hépatique catalysant l’oxydation de l’acide homogentisique.
C’est à partir de ces études qu’il créa le concept errors inborn of metabolism.
Parallèlement, en 1866, Virchow observa chez un homme de 67 ans une pigmentation très particulière de certains tissus ayant, à l’oeil, une couleur gris-bleu et au microscope une couleur ocre.
C’est à partir de cette observation qu’il créa le terme d’ochronose, mais ce n’est que près de 40 ans plus tard qu’Albrecht reconnut qu’alcaptonurie et ochronose résultaient d’une même cause.
En 1950, Suda et Takeda isolèrent une enzyme à partir d’une souche bactérienne (Pseudomonas) intervenant dans le métabolisme de la tyrosine, et l’appelèrent homogentisicase.
Ils étudièrent simultanément les propriétés d’une enzyme voisine provenant de foie de lapin.
Étude clinique :
A – Alcaptonurie :
Les urines des malades atteints d’alcaptonurie noircissent à l’air.
En fait, beaucoup plus souvent, les malades n’ont noté aucune anomalie de coloration de leurs urines pendant l’enfance et l’adolescence, le diagnostic n’étant évoqué qu’en présence des arthropathies ochronotiques, voire au cours d’une intervention devant la coloration gris-bleu du cartilage articulaire.
En effet, les malades alcaptonuriques, même s’ils ont des arthropathies ochronotiques, émettent des urines de couleur normale qui ne noircissent pas, même après plusieurs heures, si leur pH reste acide.
Ce n’est que lorsqu’elles sont alcalines que le noircissement se produit en quelques minutes, de la surface à la profondeur.
Une méthode de dosage enzymatique spécifique permet le dosage quantitatif de l’acide homogentisique dans l’urine mais aussi dans le sérum et dans divers tissus.
Il existe aussi un dosage colorimétrique de l’acide homogentisique ainsi que par chromatographie liquide à haute pression (HPLC).
B – Ochronose :
Les premières manifestations cliniques de l’ochronose sont tardives, vers 20 ou 30 ans.
Il s’agit généralement de la pigmentation gris-bleu de la sclère ou du pavillon de l’oreille.
La pigmentation de la sclère siège habituellement à mi-chemin entre le bord de la cornée et l’angle interne ou externe de l’oeil ; à l’oreille elle débute sur le pavillon et l’anthélix puis s’étend au tragus.
Le cérumen peut aussi prendre une coloration noirâtre.
Dans le creux axillaire et dans les régions génitales, la peau peut prendre aussi une coloration brunâtre.
Généralement, la pigmentation des tissus superficiels est discrète et reste parfois méconnue pendant plusieurs mois, voire plusieurs années.
La pigmentation des cartilages et fibrocartilages est souvent intense, de couleur noirâtre et très étendue.
La pigmentation du cartilage articulaire débute dans les couches les plus profondes.
Microscopiquement, les dépôts de pigments dans la synoviale et le cartilage articulaire sont intra- et extracellulaires mais on peut aussi trouver des dépôts de pigments dans les disques intervertébraux, le cartilage trachéal, les cellules épithéliales tubulaires rénales, le pancréas, les parois des artères et artérioles.
En microscopie électronique, le pigment est décelé à la surface des fibres collagènes.
Dans la synoviale, on observe, surtout à sa partie superficielle, des fragments de cartilage pigmentés avec au maximum la formation de nodules polypoïdes noirâtres résultant probablement d’une réaction à corps étrangers.
Ces nodules sont dénommés corps ostéochondraux par R Schumacher.
Le pigment ochronotique est probablement un polymère de l’acide homogentisique mais sa constitution exacte n’est pas connue.
Son mécanisme de formation est probablement identique à celui du noircissement de l’urine, c’est-à-dire oxydatif et non enzymatique.
C – Spondylarthropathie ochronotique :
L’atteinte rachidienne se signale cliniquement par un enraidissement plus ou moins douloureux du rachis lombaire et à un moindre degré cervical.
Au fil des années et plus tardivement, la lordose lombaire s’efface et souvent est remplacée par un certain degré de cyphose.
La taille peut d’ailleurs diminuer de quelques centimètres.
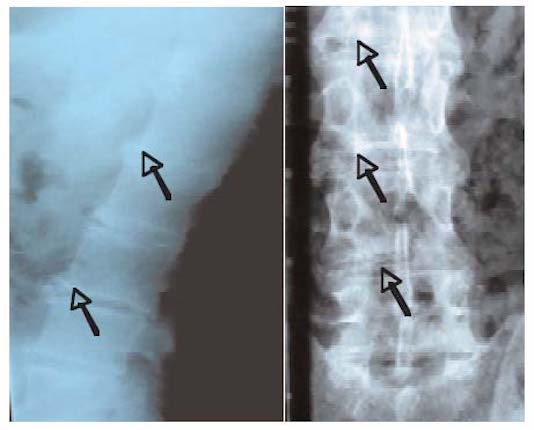
Radiologiquement, le signe le plus caractéristique est la calcification des disques intervertébraux particulièrement étendue avec progressivement apparition d’un important affaissement discal avec parfois phénomène du vide discal et au maximum fusion vertébrale.
Ce sont ces discopathies étendues et non des tassements vertébraux qui sont responsables de la diminution de la taille de ces patients.
En revanche, l’ostéophytose est généralement modérée et les sacro-iliaques ne sont pas atteintes.
Dans les formes évoluées, les altérations rachidiennes rappellent les déformations observées dans les formes sévères de spondylarthrite ankylosante.
D – Arthropathies ochronotiques :
Les arthropathies ochronotiques sont généralement plus tardives que l’atteinte rachidienne.
Elles semblent plus sévères chez l’homme bien que le sex-ratio soit de 1.
Les articulations le plus souvent atteintes sont les genoux, puis les épaules et les hanches.
En revanche, les petites articulations ne sont presque jamais atteintes.
Les arthropathies se signalent cliniquement par un enraidissement plus ou moins douloureux, avec assez souvent au genou, un flessum de quelques degrés, et parfois un épanchement synovial dont le compte cellulaire évoque une arthropathie mécanique (moins de 1 000 cellules/mm3).
Il est à noter que le liquide synovial ne noircit pas après alcalinisation.
Rarement, l’arthropathie ochronotique donne lieu à une inflammation aiguë pseudogoutteuse, et on a pu alors noter la présence de cristaux de pyrophosphate de calcium.
Les signes radiologiques ne sont visibles que plus tardivement, quelques mois, voire quelques années après les premières manifestations cliniques.
Ils n’ont aucun caractère spécifique et ressemblent trait pour trait aux lésions arthrosiques : amincissement de l’interligne articulaire, condensation osseuse sous-chondrale, ostéophytose. Parfois, le diagnostic d’arthropathie ochronotique est évoqué lors d’arthroscopies devant la pigmentation noirâtre du cartilage articulaire.
E – Autres manifestations ochronotiques :
Un souffle systolique a été observé dans 15 à 20 % des cas, probablement en rapport avec des dépôts de pigment ochronotique.
Les valvulopathies mitrales et/ou aortiques sont en effet dues partiellement à des dépôts de pigment.
Les dépôts de pigments siègent parfois dans la prostate et sont rendus responsables de dysurie pouvant entraîner une sanction chirurgicale.
La rupture du tendon d’Achille imprégné de pigment ochronotique a été signalée.
Physiopathologie de l’ochronose :
A – Synthèse et dégradation de l’acide homogentisique :
Chez les mammifères, la majeure partie de la phénylalanine et de la tyrosine alimentaire est oxydée en acide acétoacétique après une cascade de transformations enzymatiques.
La transformation de l’acide homogentisique en acide maléylacétoacétique étant catabolisée par l’acide homogentisique-oxydase.
Cette enzyme est largement répandue dans les tissus mais surtout dans le foie.
L’activité de l’enzyme purifiée est à présent bien connue. Son activité s’exerce à un pH optimal de 7 environ.
Elle nécessite la présence spécifique d’ions ferreux (les autres métaux divalents n’ont pas d’action) comme la plupart des autres oxygénases intervenant dans les réactions d’ouverture du noyau aromatique des acides aminés.
B – Physiopathologie des lésions articulaires :
La physiopathologie des lésions articulaires est mal connue.
L’acide homogentisique, ou plus exactement le pigment ochronotique, modifie probablement la structure du cartilage et provoque des lésions dégénératives proches de celles de l’arthrose.
L’une des plus caractéristiques est la perte de la striation des fibres de collagène suivie de leur gonflement et de leur rupture.
Angeles et al ont montré que la croissance des chondrocytes de lapin et des chondrocytes humains en culture cellulaire monocouche était inhibée par l’acide homogentisique proportionnellement à sa concentration dans le milieu de culture.
L’injection d’acide homogentisique dans les genoux de lapin provoque des lésions cartilagineuses ressemblant d’assez près à celles de l’arthrose.
D’ailleurs l’acide homogentisique provoque une inhibition de la lysilhydroxylase, du moins chez l’embryon de poulet.
Or l’hydroxylysine produite par cette enzyme joue un rôle majeur dans le pontage des fibrilles du collagène de type II.
C’est d’ailleurs en maintenant l’ion fer dans sa forme réduite que l’acide ascorbique augmente la transformation oxydative de l’acide homogentisique, et c’est pourquoi il a été un instant proposé dans le traitement de l’alcaptonurie, malheureusement sans succès.
C’est par une analyse systématique de l’activité des enzymes intervenant successivement dans le métabolisme de la tyrosine que La Du et al démontrèrent, il y a 40 ans, que seule l’activité de l’acide homogentisiqueoxydase est absente dans l’alcaptonurie, alors que les autres enzymes ont une activité normale, et que l’absence d’activité est liée soit à un défaut spécifique de production de l’enzyme, soit à une altération de sa structure la rendant inactive.
Enfin, il est intéressant de noter que l’acide homogentisique est non seulement filtré par le glomérule mais aussi sécrété par les tubes puisque sa clairance est de 400 à 500 mL /min.
Cette excrétion urinaire massive explique qu’il soit nécessaire d’attendre plusieurs années avant que les dépôts ochronotiques finissent par s’accumuler.
Génétique :
Osler découvrit pour la première fois chez deux frères alcaptonuriques une pigmentation de la sclère et des oreilles qu’il rattacha à une même anomalie métabolique.
Garrod en 1902 décrivit, rappelons-le, le concept de maladie héréditaire à partir d’observations de familles d’alcaptonuriques.
Il suggéra même que l’enzymopathie était de transmission autosomale récessive, notamment en raison de sa fréquence dans les familles issues de mariages consanguins.
Ce mode de transmission a été confirmé depuis par plusieurs enquêtes.
La prévalence de l’alcaptonurie est estimée à 3-5 par million, et son apparition remonte au moins à 3 000 ans.
Récemment plusieurs équipes ont montré que la maladie est transmise par un seul gène localisé sur une région de 16 centimorgan sur le bras long du chromosome 3.
Traitement :
Il n’y a aucun traitement efficace des arthropathies ochronotiques.
Comme dans les arthropathies dégénératives, on peut avoir recours avec succès aux arthroplasties.
Une thérapie génique visant à pallier le défaut d’homogentisicase permettra peut-être un jour de guérir cette erreur innée du métabolisme pour reprendre le fameux concept élaboré pour la première fois par Garrod.







")

")

{kind=link}