



















Introduction :
Sous le terme de neurofibromatoses (NF) sont regroupées des maladies distinctes n’ayant en commun le plus souvent que certains signes cutanés : taches pigmentaires et tumeurs.
Toutes ces affections ont longtemps été désignées sous le nom de maladie de von Recklinghausen.
Le terme de maladie de von Recklinghausen ne désigne maintenant que la plus fréquente des neurofibromatoses, la NF1.
Au moins deux maladies bien identifiées à transmission autosomique dominante font partie de ce groupe hétérogène, la NF1 et la NF2.
Nosologie :
A – HISTORIQUE NOSOLOGIQUE :
 C’est au XIIIe siècle que le premier malade atteint de NF est décrit dans un manuscrit hongrois par le moine Henricus.
C’est au XIIIe siècle que le premier malade atteint de NF est décrit dans un manuscrit hongrois par le moine Henricus.
Au XVIe, Aldrovandi de Bologne, dans son ouvrage Monstorum Historia, décrit une tumeur spécifique de la NF1, le neurofibrome plexiforme.
Au XVIIIe siècle, Buffon dans son Histoire Naturelle rapporte le cas d’un enfant ayant des tumeurs cutanées et des taches pigmentées. Cruveilhier, dans son Anatomie Pathologique du corps donne le premier une description précise de la NF1.
Plus tard, Akenside, Ludwig et Tilesius en décrivent différents aspects et reconnaissent la possibilité de transmission familiale.
Smith en 1849, Hitchcock en 1862, von Recklinghausen en 1882, en précisent la description.
Les « neurinomes acoustiques », aujourd’hui nommés schwannomes vestibulaires, la manifestation majeure de la NF2, sont décrits par Wishart (1822) et Knoblauch (1843), puis Smith et von Recklinghausen, et enfin Henneberg et Koch en 1903.
En 1953, Schull et Neel précisent l’importance diagnostique des troubles pigmentaires dans ce que l’on pense toujours n’être qu’une seule affection nommée maladie de von Recklinghausen et qui regroupe en fait au moins deux maladies, la NF1 et la NF2. Waardenbourg puis Lisch signalent la fréquence des nodules iriens.
La confusion nosologique se poursuit jusqu’en 1970, les travaux de Young, Eldridge et Gardner puis ceux de Riccardi et Mulvihill établissent définitivement la séparation des deux entités, la NF1 et la NF2. À la fin des années 1980, le cas de John Merrick, elephant man, longtemps considéré comme une NF1, est rattaché au syndrome de Protée.
B – NOSOLOGIE MODERNE :
Depuis la conférence de consensus du National Institute of Health (NIH) de 1988, les critères diagnostiques de NF1 et NF2 ont été précisés.
La classification de Riccardi avait l’ambition de distinguer d’autres tableaux cliniques mal adaptés aux cadres précédents.
La validité des critères diagnostiques de la conférence de consensus du NIH a été confirmée par la biologie moléculaire qui a permis d’identifier deux gènes distincts pour la NF1 et la NF2 : le gène NF1 est situé sur le chromosome 17 et le gène NF2 sur le chromosome 22.
Neurofibromatose 1 ou maladie de von Recklinghausen :
A – ÉPIDÉMIOLOGIE :
La NF1 est la plus fréquente des maladies autosomiques dominantes, avec une incidence d’environ une naissance sur 3 000 à 3 500.
Elle représente 95 % de l’ensemble des NF. Sa prévalence est d’environ un individu sur 4 000.
Elle se transmet sur un mode autosomique dominant sans aucune prédilection ethnique.
La moitié des cas sont sporadiques, traduisant la fréquence des mutations spontanées.
La variation phénotypique est importante, même au sein d’une même famille.
Sa morbidité et sa mortalité sont liées à la survenue de complications multisystémiques, tumeurs cérébrales, tumeurs malignes des gaines nerveuses (TMGN) et vasculopathies.
B – GÈNE NF1 :
Le gène NF1 a été identifié par clonage positionnel en 1990.
Il s’étend sur plus de 350 kilobases dans la région péricentromérique du chromosome 17 (17q11.2) et est composé de 59 exons.
Il s’agit d’un gène de grande taille dont l’exploration est difficile, et ce d’autant plus qu’il existe des séquences homologues localisées sur les chromosomes 15, 21 et 22.
La pénétrance du gène est proche de 100 % à 5 ans.
Le produit du gène NF1, la neurofibromine, est une protéine cytoplasmique composée de 2 818 acides aminés incluant une région de 360 acides aminés (domaine GRD) similaire au domaine catalytique de type GTPase des protéines p120GAP de mammifères, IRA1, IRA2 et sar1 de levure (Saccharomyces cerevisiae et S. pombe) et GAP1 de drosophile.
La fonction commune de ces molécules est de stimuler la conversion de la forme active de la protéine p21ras liée au GTP en une forme inactive liée au GDP.
La neurofibromine intervient donc très probablement dans le contrôle de la différenciation et de la prolifération cellulaire.
Cependant, les données actuelles ne permettent pas de définir de façon formelle si ce contrôle se situe en amont et/ou en aval de l’activation de p21ras.
En effet, certains résultats laissent à penser que la neurofibromine pourrait être, selon le type cellulaire, un régulateur négatif ou un effecteur direct de p21ras.
Il existe plusieurs isoformes de la neurofibromine liées à des phénomènes d’épissage alternatif.
L’isoforme II, caractérisée par l’insertion de 21 acides aminés au sein du domaine GRD, possède une distribution tissulaire proche de celle de l’isoforme I.
En revanche, l’isoforme III, caractérisée par l’insertion de 18 acides aminés dans la partie carboxyterminale de la protéine, est exprimée de façon préférentielle dans les muscles cardiaque, squelettiques et lisses.
Il existe une quatrième isoforme résultant de l’insertion de dix acides aminés entre les résidus 420 et 421 de la neurofibromine et dont l’expression semble être restreinte dans le système nerveux central.
Environ 5 % des patients atteints de NF1 développent des tumeurs malignes suggérant que le gène NF1 est un gène suppresseur de tumeur, ce qui a été récemment démontré.
Par ailleurs et conformément à l’hypothèse de Knudson, des pertes d’hétérozygotie du gène NF1 ont été retrouvées dans la plupart des types de tumeurs développées par les patients présentant une NF1 : TMGN (anciens neurofibrosarcomes), schwannomes, syndromes myélodysplasiques, phéochromocytomes et même tout récemment neurofibromes bénins.
Il existe de grandes variations phénotypiques inter- et intrafamiliales dans la NF1.
Les critères diagnostiques majeurs de la NF1, par exemple les taches café au lait (TCL), les neurofibromes cutanés et les nodules de Lisch sont présents dans plus de 90 % des cas à la puberté, mais le nombre des lésions varie énormément d’un individu à l’autre.
En revanche, certaines manifestations de la NF1 ne surviennent que chez une minorité de malades ; c’est le cas des difficultés d’apprentissage, des gliomes des voies optiques, des crises comitiales, des pseudarthroses et des scolioses, des TMGN et des phéochromocytomes.
Une minorité significative de malades, environ 15 à 20 %, souffre d’une morbidité importante liée à la NF1, alors qu’une majorité n’est atteinte que de formes modérées.
Certaines de ces variations d’expression phénotypique pourraient refléter différentes mutations sur le gène NF1, bien que jusqu’à présent les tentatives de corrélation génotype-phénotype se soient révélées infructueuses.
Seules des délétions importantes du gène NF1 ont pu être associées à un phénotype particulier.
Par ailleurs, plusieurs groupes tentent actuellement de caractériser les bases moléculaires des associations NF1-syndrome de Noonan.
En effet, les données actuelles ne permettent pas de définir si cette association est une forme allélique de NF1, comme cela semble être le cas pour le syndrome de Watson, ou si elle est liée à l’existence de deux mutations distinctes situées sur des gènes génétiquement liés (gènes contigus).
Néanmoins, il est établi que ces variations dans la mutation NF1 ne sont pas la seule source de variations phénotypiques.
Il existe, en effet, également une différence d’expression considérable au sein d’une même famille.
La variation phénotypique interindividuelle pourrait être liée à des gènes modificateurs et/ou des facteurs environnementaux.
Les gènes modificateurs ne sont pas situés sur le locus NF1, mais leur activité ou leur répression conduirait à la modification de l’expression phénotypique.
Cette hypothèse semble actuellement établie.
Le nombre de TCL, de NF, la survenue ou non de gliomes optiques, d’une scoliose, d’une épilepsie, ou de difficultés d’apprentissage semblent dépendants de gènes modificateurs.
C – DIAGNOSTIC CLINIQUE :
1- Signes cardinaux dermatologiques :
* Taches café au lait :
Les TCL sont les premières manifestations de la NF1.
Elles sont souvent congénitales et apparaissent rarement après l’âge de 2 ans. Leur répartition est aléatoire, leur contours sont nettement tracés et leur teinte marron plus ou moins foncé parfois à la limite de la visibilité.
Le diamètre des TCL varie de 0,5 à 50 cm, mais la majorité d’entre elles mesurent moins de 10 cm. Les anomalies histologiques se résument à une forte hyperpigmentation en foyer de la membrane basale.
En microscopie électronique, les mélanosomes géants sont très fréquents, mais sont aujourd’hui considérés comme peu spécifiques.
De même, l’augmentation des terminaisons nerveuses observée plus récemment dans les TCL n’est pas spécifique de la NF1 mais retrouvée également dans les nævus géants congénitaux.
À l’adolescence, les TCL sont présentes dans plus de 90 % des cas.
Au cours de la vie adulte, les TCL deviennent souvent plus pâles, peu visibles, et certaines disparaissent. Les TCL constituent un des meilleurs signes diagnostiques de NF1, presque toujours présentes avant l’âge de 5 ans.
La taille et le nombre de TCL sont importants à considérer : les TCL de taille supérieure à 0,5 cm dans l’enfance et d’au moins 1,5 cm après la puberté ont valeur diagnostique à condition d’être en nombre supérieur ou égal à six (plus de 25 % des enfants d’âge scolaire et 14 % des adultes ont une à cinq TCL).
* Lentigines :
Les lentigines ont l’aspect de TCL de petite taille, macules de 1 à 3 mm de diamètre qui siègent de manière élective dans les plis axillaires où leur spécificité est la plus grande, dans les plis inguinaux et sous-mammaires.
Elles peuvent toucher la nuque, l’espace sous-mentonnier ou encore être diffuses.
Rarement présentes avant l’âge de 2 ans, on les retrouve dans 80 % des cas après la puberté.
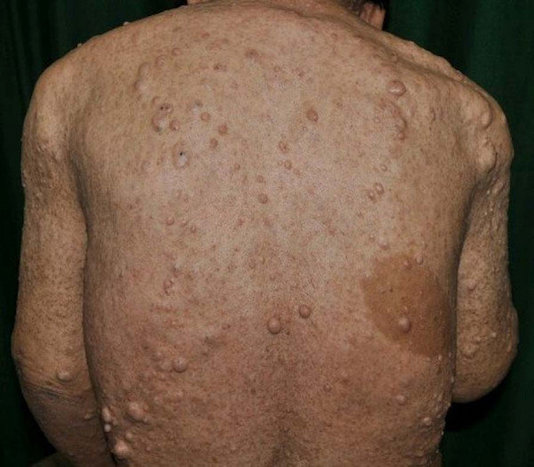
* Neurofibromes :
Les neurofibromes sont des tumeurs bénignes.
Ils se développent dans et le long des nerfs et des gaines nerveuses.
Leur présence est un signe cardinal diagnostique de NF1.
Les neurofibromes cutanés se développent dans le derme et l’épiderme.
Ce sont des petites tumeurs molles, mobiles avec la peau, sessiles ou pédiculées à type de molluscum pendulum.
De couleur chair, rosée ou violacée, leur consistance est particulière, élastique et dépressible.
Les neurofibromes cutanés n’apparaissent qu’à la puberté et sont exceptionnellement absents à l’âge adulte (95 % des adultes en sont atteints).
Les neurofibromes varient en taille, de 0,1 cm à quelques centimètres, et en nombre, de quelques uns à plusieurs milliers.
En l’absence de traitement, ils peuvent atteindre une taille importante, en surface et relief (aspect en « battant de cloche », en « besace »).
Ils augmentent parfois en nombre et en taille pendant la grossesse.
Si le tronc est leur siège principal, toutes les autres parties du corps peuvent être atteintes (face, membres incluant mains et pieds, cuir chevelu).
L’atteinte des aréoles mammaires chez la femme est particulièrement fréquente (80 % des femmes adultes).
Ces neurofibromes peuvent être prurigineux, notamment au moment de leur apparition, et parfois douloureux quand ils ont une composante sous-cutanée.
Les neurofibromes nodulaires périphériques ou neurofibromes souscutanés sont présents à l’âge adulte chez environ 20 % des malades. Ils apparaissent rarement avant la seconde enfance.
Ils se développent à partir de troncs nerveux plus importants que les neurofibromes cutanés.
Ils sont plus palpables que visibles, bombant sous la peau, sphériques ou ovoïdes, isolés ou en « chapelet », fermes, toujours sensibles ou douloureux à la pression qui peut provoquer des paresthésies sur le trajet nerveux à distance.
Les neurofibromes plexiformes diffus étaient autrefois nommés dans leur forme importante « névrome plexiforme » ou « tumeur royale ».
Ils sont distincts histologiquement et cliniquement des neurofibromes dermiques ; ils impliquent toutes les couches de la peau et peuvent pénétrer profondément muscle, os et viscères.
Ce sont des tuméfactions cutanées et sous-cutanées de taille très variable (de quelques centimètres à plusieurs dizaines de centimètres, voire étendues à tout un segment corporel).
La peau en regard est toujours anormale, mélange d’hypertrophie, d’hypertrichose et de pigmentation marron souvent proche de celle d’une TCL.
Ils sont le plus souvent mous, de texture irrégulière, ayant parfois un aspect de cutis laxa localisé.
Ils siègent surtout sur le tronc, la tête, les membres et le cou. Histologiquement, la croissance intrafasciculaire des cellules tumorales conduit à une tuméfaction du nerf ; la composante schwannienne est importante ; la tumeur est mal limitée en travées irrégulières.
Les neurofibromes plexiformes sont souvent congénitaux ; ils sont toujours visibles avant 5 ans. Retrouvés dans un tiers des cas, ils tendent à se développer à partir de l’adolescence.
Leur retentissement esthétique peut être considérable.
Les neurofibromes plexiformes nodulaires sont moins fréquents.
Ils ont l’aspect de multiples neurofibromes sous-cutanés, regroupés en grappes, en cordons diposés le long de troncs nerveux.
Ils sont fermes, sensibles ou douloureux, comme les simples neurofibromes sous-cutanés isolés dont ils constituent les formes majeures.
Développés en réseau sous-cutané à partir des racines nerveuses, ces neurofibromes plexiformes nodulaires peuvent être superficiels ou profonds, à risque alors de compressions sévères (moelle, médiastin…).
2- Autres signes cardinaux :
* Nodules de Lisch :
Les nodules de Lisch sont de petits hamartomes iriens qui n’entraînent aucun trouble de la fonction visuelle.
Ils constituent un critère diagnostique de NF1 (au moins deux nodules dans chaque champ).
Leur taille et leur nombre augmentent avec l’âge : retrouvés seulement chez 10 % des sujets avant 6 ans, ils sont présents dans plus de 90 % des cas après 16 ans.
Ils sont parfois visibles à l’ophtalmoscope, voire à l’oeil nu, mais leur recherche nécessite un examen minutieux à la lampe à fente fait par un ophtalmologue averti.
Ils ont l’aspect de petits nodules jaune-brun le plus souvent, parfois très pâles, en saillie sur la face antérieure de l’iris.
Ces hamartomes pigmentés sont faits de cellules fusiformes recouvrant un groupement de cellules næviques.
Exceptionnellement décrits en dehors de la NF1, ils sont quasiment pathognomoniques de cette affection.
* Gliome des voies optiques :
Le gliome des voies optiques est la tumeur intracérébrale la plus fréquente au cours de la NF1.
Son incidence estimée varie de 1,5 à 15 % en fonction du mode de recrutement.
L’incidence réelle du gliome des voies optiques symptomatique au cours de la NF1 est probablement comprise entre 1,5 et 7,5 %.
Touchant surtout nerfs et/ou chiasma optique, il peut s’étendre le long des voies optiques.
Il n’est symptomatique que dans 50 % des NF1.
Sa recherche dans un but diagnostique est rarement nécessaire, même avant 6 ans où l’examen ophtalmologique est souvent peu fiable.
À l’inverse, la découverte d’un gliome des voies optiques commande une enquête diagnostique de NF1 (25 % des gliomes des voies optiques sont associés à une NF1).
* Atteintes osseuses spécifiques :
+ Dysplasies des os longs :
Les dysplasies des os longs sont congénitales, atteignant préférentiellement le tibia.
Les manifestations cliniques peuvent être précoces (courbure congénitale d’une jambe) ou n’apparaître qu’à la marche.
Ces dysplasies sont souvent révélées par des fractures avec pseudarthrose secondaire.
+ Dysplasies des ailes sphénoïdes :
Les dysplasies des ailes sphénoïdes sont congénitales, en général unilatérales et non évolutives.
Elles sont souvent accompagnées d’un neurofibrome plexiforme orbitaire.
Elles sont rares, moins de 1 % des cas de NF1, mais très évocatrices.
Les conséquences esthétiques de ces dysplasies peuvent être importantes : asymétrie faciale, exophtalmie ou énophtalmie.
+ Dysplasies vertébrales :
Les dysplasies vertébrales les plus caractéristiques sont l’accentuation de la concavité postérieure (ou antérieure) de certains corps vertébraux (aspect en « feston » ou scalloping), un amincissement des pédicules, un élargissement des trous de conjugaison. C’est un signe très évocateur de NF1.
3- Démarche diagnostique :
Chez l’adulte, le diagnostic de NF1 est en règle facile sur les données de l’examen clinique : la NF1 est une maladie à critères (au nombre de sept dont trois sont des signes dermatologiques fréquemment retrouvés).
Le diagnostic est établi chez un individu s’il présente deux critères ou plus.
Dans la petite enfance, où les TCL peuvent demeurer longtemps le seul signe, et en l’absence d’antécédents familiaux de NF1, le diagnostic demeure parfois en suspens.
Dans cette période de la vie, l’examen ophtalmologique à la recherche de nodules de Lisch est peu rentable ; l’emploi de l’imagerie par résonance magnétique (IRM) est rarement justifié, mais les radiographies des os longs peuvent être un complément utile au diagnostic.
La pénétrance de la maladie est complète à l’âge de 5 ans.
Le diagnostic moléculaire, parfois possible, est exceptionnellement nécessaire.
Il peut être fait dans les formes familiales par analyse de ségrégation des polymorphismes de l’acide désoxyribonucléique.
Dans les formes sporadiques, seule la mise en évidence de la mutation peut permettre le diagnostic ; les techniques à notre disposition actuellement ne permettent de caractériser que 20 à 25 % des anomalies moléculaires.
Ces performances médiocres devraient être améliorées dans les années à venir.
Le diagnostic anténatal n’est envisageable dans les formes familiales que si les analyses de ségrégation sont informatives, et beaucoup plus rarement dans les formes sporadiques quand la mutation a été identifiée.
Dans ces cas, une biopsie de villosités choriales peut être faite vers les dixième ou onzième semaines de gestation et l’absence ou la présence de transmission du gène NF1 muté authentifiées dans les cellules embryonnaires.
La biopsie de villosités choriales entraîne un faible risque d’avortement.
Le diagnostic anténatal n’est réalisé que si les parents envisagent d’interrompre la grossesse dans le cas où l’embryon serait porteur du gène NF1 muté.
Outre d’éventuelles oppositions personnelles d’ordre moral, surtout religieux, le recours au diagnostic anténatal reste très limité, surtout du fait de l’impossibilité de prédiction de la gravité phénotypique de la forme éventuellement transmise : de 15 à 20 % des malades sont gravement touchés par la mutation NF1.
Le diagnostic préimplantatoire n’est pas à l’ordre du jour.
D – COMPLICATIONS :
1- Manifestations orthopédiques :
Les cyphoscolioses sont fréquentes.
Le plus souvent, il s’agit d’atteintes discrètes, peu évolutives, non spécifiques, sans dystrophie osseuse associée et contrôlables par des méthodes de rééducation fonctionnelle ou de contention.
Les scolioses majeures sont rares, moins de 5 % des cas, dues à des dystrophies vertébrales plus ou moins étendues (déformation des corps vertébraux, amincissement des pédicules, élargissement du canal rachidien), avec ou sans dystrophie costale.
Elles peuvent avoir un retentissement cardiorespiratoire important et être associées à des méningocèles ou à des neurofibromes nodulaires para- ou intravertébraux faisant risquer une compression médullaire.
Ces troubles de la statique vertébrale apparaissent surtout dans la petite enfance.
Les manifestations majeures nécessitent un traitement chirurgical.
Les pseudarthroses touchent de 1 à 3% des sujets atteints de NF1.
Leur traitement, complexe, long et difficile, conduit souvent après des années d’effort à une amputation.
L’introduction de techniques de microchirurgie (greffons vascularisés) en a amélioré le pronostic. Une déformation du sternum, pectus excavatum ou carinatum, est rapportée dans 2 % des cas.
La macrocéphalie est fréquente en l’absence d’anomalies structurales ou de troubles fonctionnels cérébraux, ainsi qu’une petite taille.
2- Manifestations et complications neurologiques :
Des astrocytomes de différents types, le plus souvent pilocytiques, peuvent se développer, volontiers sur la ligne médiane, parfois dans la fosse postérieure, les hémisphères cérébraux ou la moelle.
Ils sont considérés généralement comme peu ou pas évolutifs.
Cependant, certains peuvent entraîner des troubles sévères et évoluer vers l’astrocytome anaplasique.
L’hydrocéphalie, présente dans 2 % des cas, est le plus souvent secondaire à une sténose de l’aqueduc de Sylvius, sans tumeur identifiable.
Des vasculopathies, sténoses, anévrismes, ont été rapportés.
L’épilepsie semble augmentée en fréquence.
Des céphalées existent chez une grande proportion de malades, souvent migraineuses.
Les neurofibromes nodulaires sont parfois multiples et massifs, causes de graves compressions médullaires (en cas de localisation intradurale) ou nerveuses périphériques (radiculaires, plexiques).
Les difficultés d’apprentissage constituent un problème majeur au cours de la NF1.
Elles sont remarquables par leur fréquence, de 30 à 40 % des cas, et par leurs aspects atypiques.
Ces troubles neurocognitifs altèrent parfois considérablement la scolarité, alors que les retards mentaux proprement dits sont de fréquence comparable à celle de la population générale.
Le tableau est souvent proche d’un syndrome de déficit de l’attention avec ou sans hyperactivité : troubles de l’attention, difficultés de coordination motrice, déficit de la mémoire récente, troubles perceptifs surtout visuels entraînant des difficultés à dessiner, écrire, lire, calculer, à réaliser cartes et diagrammes, à se repérer dans l’espace, à déchiffrer les expressions gestuelles ; des difficultés d’élocution, troubles dysarthriques mais aussi prosodiques, sont souvent associés.
Sur le plan pratique, il est important que les parents et les enseignants soient informés dès la maternelle sur les difficultés d’apprentissage afin de proposer précocement une évaluation et une aide adaptées à chaque cas.
Les IRM cérébrales des sujets jeunes atteints de NF1 montrent dans 50 à 70 % des cas des hypersignaux en T2 : zones bien circonscrites, hyperintenses sans effet de masse, objets brillants non identifiés.
Ces anomalies tendent à s’estomper à l’âge adulte.
Leur signification est incertaine ; hormis une association possible de ces images à des troubles cognitifs, aucune autre anomalie clinique n’a jusque-là été signalée.
Les images cérébrales sont parfois difficiles d’interprétation et la différenciation avec une tumeur cérébrale est parfois délicate.
3- Manifestations et complications endocriniennes :
Un phéochromocytome, le plus souvent surrénalien, est présent dans moins de 1 % des cas à l’âge adulte et exceptionnellement dans l’enfance.
Le phéochromocytome est souvent isolé ou exceptionnellement peut s’intégrer dans un syndrome de néoplasies endocriniennes multiples, toucher une ou les deux glandes surrénales.
Rarement, le phéochromocytome siège sur la bifurcation aortique ou dans le médiastin.
Exceptionnellement asymptomatique, il s’accompagne le plus souvent de signes cliniques intermittents : crises hypertensives mêlant poussées sudorales, anxiété, agitation, céphalée, palpitations.
L’hypertension peut être permanente.
La rareté du phéochromocytome au cours de la NF1 et son caractère exceptionnellement asymptomatique rendent injustifié le dosage systématique des catécholamines.
Les anomalies pubertaires sont rares, soit puberté précoce associée à un gliome du chiasma, soit retards pubertaires dans 1,5 % des cas.
La puberté précoce révèle un tiers des gliomes des voies optiques.
Des pubertés précoces sans atteinte des voies optiques sont possibles.
4- Manifestations et complications ophtalmologiques :
La principale complication ophtalmologique de la NF1 est le gliome des voies optiques.
Environ 50 % des patients ont des anomalies de l’examen ophtalmologique au moment du diagnostic de gliome.
Il s’agit le plus souvent d’une baisse de l’acuité visuelle uni- ou bilatérale, plus rarement d’une exophtalmie.
Ces anomalies peuvent être asymptomatiques.
Il faut insister en particulier sur la difficulté du diagnostic de baisse de l’acuité visuelle chez le jeune enfant.
Les signes endocrinologiques sont plus rares (puberté précoce) et sont associés aux gliomes de localisation chiasmatique.
L’histoire naturelle des gliomes des voies optiques au cours de la NF1 est très variable : il est rare d’observer une évolution clinique ou radiologique au cours du suivi de ces tumeurs, même si la lésion est cliniquement symptomatique au moment du diagnostic.
Toutefois, les évolutions rapides mettant en jeu le pronostic visuel et vital sont possibles et totalement imprévisibles.
La période à risque d’apparition des complications est la petite enfance, mais la surveillance clinique (ophtalmologique, neurologique et endocrinologique) de ces patients doit être prolongée. Une action thérapeutique ne s’envisage qu’en cas de gliome des voies optiques symptomatique et évolutif.
De nombreuses anomalies peuvent être retrouvées au cours de la NF1 : lésions choroïdiennes hamartomateuses (de 35 à 50 % des cas) ; hypertrophie des nerfs cornéens (15 % des cas) ; ptôse palpébrale isolée (9 % des cas) ou associée à un neurofibrome palpébral ou orbitaire ; anomalies de la convergence ; glaucome congénital (0,5 % des cas), souvent associé à un neurofibrome plexiforme palpébral.
5- Atteintes artérielles :
Les dysplasies artérielles pariétales fibromusculaires sont fréquentes et peuvent toucher l’aorte, les artères mésentériques, pulmonaires, cérébrales et rénales.
Cette dernière localisation est souvent révélée par une hypertension.
C’est avec le phéochromocytome surrénalien la seconde cause d’hypertension artérielle au cours de la NF1.
Elle est accessible parfois à une angioplastie endoluminale percutanée.
Des vasculopathies induites par la radiothérapie chez les enfants atteints de NF1 et de gliomes des voies optiques ont été rapportées, témoignant d’une susceptibilité particulière.
6- Atteintes gastro-intestinales :
En plus des tumeurs carcinoïdes, des neurofibromes et d’autres tumeurs du tractus digestif ont été rapportés au cours de la NF1.
La fréquence des neurofibromes digestifs a été estimée à environ 2 % des malades atteints de NF1 ; leur localisation la plus fréquente est le jéjunum.
Des léiomyomes, des ganglioneuromes et des sarcomes ont également été rapportés.
Les symptômes associés sont des douleurs, une dyspepsie et une constipation. Hématémèse et méléna peuvent être révélateurs.
7- Complications pulmonaires :
Les neurofibromes intrapulmonaires sont généralement asymptomatiques.
Toutefois, exceptionnellement, des neurofibromes de taille importante peuvent se révéler par l’apparition d’une toux ou de difficultés respiratoires.
L’existence d’une scoliose importante entraîne le plus souvent une insuffisance respiratoire restrictive par réduction des volumes respiratoires pulmonaires et fibrose pulmonaire.
Cette insuffisance respiratoire peut mettre en jeu le pronostic vital dans les cas sévères.
Les malformations vasculaires pulmonaires peuvent être à l’origine d’hémoptysies.
8- Complications du système urinaire :
Une hydronéphrose ou des troubles urinaires peuvent apparaître.
Ces symptômes résultent généralement de la compression du tractus urinaire par des neurofibromes rétropéritonéaux ou pelviens.
9- Complications esthétiques et fonctionnelles :
Les neurofibromes plexiformes congénitaux peuvent être responsables de complications esthétiques majeures (hypertrophie de segments corporels), avec parfois sur la face atteinte ophtalmologique (amblyopie).
Les neurofibromes cutanés, qui apparaissent souvent à l’adolescence, ont souvent de lourdes conséquences psychologiques et sociales par leur caractère affichant parfois spectaculaire.
Les hamartomes anémiques sont fréquents au cours de la NF1.
Des plaques rosées atrophiques du tronc, souvent préthoraciques, sont aussi parfois rencontrées, déprimées, planes, à bords nets.
Les schwannomes cutanés ou sous-cutanés sont exceptionnels au cours de la NF1.
Les xanthogranulomes juvéniles sont exceptionnels, moins de 1 % de cas de NF1.
Ils apparaissent dans les 2 premières années de la vie et régressent lentement.
Leur association avec la leucémie myéloïde juvénile chronique a été plusieurs fois rapportée.
Le risque de leucémie myéloïde est extrêmement faible au cours de la NF1 (0,004 % des cas), mais semble augmenter en cas d’association avec des xanthogranulomes juvéniles.
Le prurit, souvent contemporain de l’apparition de neurofibromes, est retrouvé dans environ 10 % des cas.
Des nappes hyperpigmentées signalent parfois la présence d’un neurofibrome plexiforme et, en situation dorsale médiane, une atteinte sous-jacente possible du névraxe.
10- Cancer et neurofibromatose 1 :
* Tumeurs malignes des gaines nerveuses :
Les TMGN, anciennement appelées neurofibrosarcomes, se développent à partir de cellules de Schwann ou fibroblastes du périnerve.
Elles représentent environ 10 % des sarcomes des tissus mous mais sont étroitement liées à la NF1. Ainsi, de 50 à 60 % des patients avec TMGN sont atteints de NF1.
La prévalence des TMGN dans les cohortes de malades atteints de NF1 est de l’ordre de 3 à 4 %.
Les TMGN surviennent au cours de la troisième décennie dans la NF1 et sont de haut grade de malignité dans 85 % des cas.
Elles se développent à partir des neurofibromes plexiformes ou des neurofibromes nodulaires.
Les signes d’appel sont les douleurs en rapport avec la masse tumorale, les signes neurologiques à type de paresthésies et l’augmentation rapide de la taille d’une tumeur généralement préexistante.
L’apparition de l’un de ces signes d’appel doit faire pratiquer une biopsie chirurgicale, répétée si le doute persiste.
* Autres cancers :
Certains autres cancers exceptionnels au cours de la NF1 ont sans doute une prévalence accrue : glioblastomes cérébraux, leucémies, rhabdomyosarcome, adénocarcinome ou tumeur carcinoïde du duodénum, phéochromocytome malin, neuroblastome et tumeur de Wilms.
11- Altération de la qualité de vie au cours de la neurofibromatose 1 :
L’imprévisibilité de l’évolution de la NF1, la survenue de complications faisant la gravité de la maladie, mais également les conséquences esthétiques et la visibilité de la maladie ont un impact significatif sur la qualité de vie des malades.
E – FORMES VARIANTES DE NEUROFIBROMATOSE 1 :
1- Syndrome de Noonan-neurofibromatose 1 :
À des signes de syndrome de Noonan (dysmorphie faciale avec cou bref, pterygium colli, ptôse palpébrale, cheveux implantés bas, oreilles implantées bas et tournées en arrière, petite taille, pectus excavatum, lymphoedème, retard pubertaire, troubles cognitifs et malformation cardiaque, surtout à type de sténose artérielle pulmonaire congénitale) sont parfois associées des manifestations évocatrices de NF1.
La génétique de cette association est discutée.
2- Syndrome de Watson :
Sont ainsi nommés des cas familiaux à transmission autosomique dominante associant TCL, sténose valvulaire pulmonaire et intelligence infranormale.
Il semble être une forme allélique de NF1.
F – ÉVOLUTION ET GRAVITÉ :
La NF1 est une affection évolutive qui, chez chaque sujet atteint, devient plus lourde année après année.
Si dans la majorité des cas la gravité des atteintes demeure limitée, tout au long de la vie des complications peuvent survenir, différentes selon les âges : neurofibromes plexiformes toujours apparents avant l’âge de 5 ans, TMGN (neurofibrosarcomes) à partir de l’adolescence.
Pour 15 à 20 % des sujets atteints de NF1, la morbidité est ou sera importante.
Chez l’enfant, les troubles de l’apprentissage et le gliome des voies optiques constituent les problèmes majeurs.
Chez l’adulte, la complication la plus redoutable est la TMGN.
L’espérance de vie des sujets atteints de NF1 est réduite d’une dizaine d’années par rapport à celle de la population générale.
Les principales causes de décès sont les néoplasies, essentiellement TMGN et tumeurs cérébrales, mais également la vasculopathie associée à la NF1 et notamment les accidents vasculaires cérébraux.
La fertilité de sujets atteints de NF1 est normale, mais des poussées de neurofibromes sont fréquentes au cours de la grossesse, ainsi que l’apparition ou l’aggravation d’une hypertension artérielle.
G – DIAGNOSTICS DIFFÉRENTIELS :
– Syndrome de McCune-Albright.
Il associe TCL à bords irréguliers, puberté précoce et dysplasie fibreuse polyostotique.
– Syndrome LEOPARD. Il associe sténose artérielle pulmonaire, lentigines multiples, petite taille et surdité.
– Syndrome de Carney.
Il associe lentigines, myxomes (en particulier cutanés et cardiaques) et anomalies endocriniennes.
– Syndrome de Protée (« elephant man »). Elephant man n’avait pas une NF1 mais un syndrome de Protée.
Ce syndrome associe de façon variable une hémihypertrophie corporelle segmentaire, une macrodactylie, des hamartomes conjonctifs et/ou épidermiques.
L’épaississement en masses cérébriformes des paumes et des plantes est particulièrement évocateur.
L’histologie des masses sous-cutanées correspond à des hamartomes lipomateux et/ou angiomateux.
La plupart des cas sont sporadiques, probablement liés à une mutation non létale, uniquement sous forme mosaïque.
– Autres affections. Nous citons le syndrome de l’hamartome épidermique, les lipomatoses et le syndrome de Bannayan-Riley-Rulvalcaba, le syndrome de Klippel-Trenaunay (hémangiome, hémihypertrophie) et les néoplasies endocriniennes multiples (coexistence de tumeurs d’au moins deux glandes endocrines fonctionnellement indépendantes).
H – SUIVI DES MALADES ATTEINTS DE NEUROFIBROMATOSE 1 :
En dehors du conseil génétique et du traitement des manifestations cutanées qui constituent la demande prioritaire des malades adultes, un suivi est nécessaire pour la détection précoce des complications de la NF1, dont beaucoup surviennent dans l’enfance.
La gravité de la NF1 est variable d’un sujet à l’autre et au sein d’une même famille.
La NF1 est une maladie dont la gravité augmente généralement avec l’âge.
Nous ne disposons d’aucun signe prédictif de l’évolution. Même devant des formes de NF1 qui paraissent bénignes, un suivi doit être proposé.
Ce suivi doit être essentiellement clinique.
Les examens effectués à titre systématique sont peu rentables pour le malade.
L’examen clinique peut facilement identifier des complications telles une scoliose, une pseudarthrose, une hypertension artérielle liée à une sténose de l’artère rénale ou à un phéochromocytome, ou encore des difficultés d’apprentissage scolaire dont le dépistage doit être le plus précoce possible.
Les examens complémentaires ne sont à effectuer que sur des arguments cliniques.
La seule exception controversée est peut-être l’IRM des voies optiques, pour la détection d’un gliome des voies optiques potentiellement agressif, en particulier chez les jeunes enfants, car l’examen ophtalmologique peut être difficile.
L’attitude suivante est proposée par les neuro- et oncopédiatres du réseau NF-France.
Chez l’enfant, lorsqu’une coopération insuffisante rend l’examen ophtalmologique difficile, soit en raison de l’âge (moins de 6 ans), soit en raison de troubles cognitifs, l’IRM cérébrale est systématique.
Si aucune anomalie n’est détectée à l’imagerie, une nouvelle IRM cérébrale est prescrite au bout de 2 ans si l’examen ophtalmologique annuel systématique reste encore incomplet.
Dès que l’âge de l’enfant le permet, une simple surveillance ophtalmologique annuelle incluant une mesure de l’acuité visuelle et un champ visuel est conseillée.
Si des anomalies sont détectées, une IRM est alors réalisée.
S’il existe une anomalie évocatrice de gliome des voies optiques à l’imagerie, seuls les gliomes des voies optiques à évolution agressive justifient une action thérapeutique.
Afin d’évaluer le potentiel agressif de la tumeur, la surveillance suivante est alors proposée : un examen ophtalmologique et une IRM tous les 3 mois pendant 6 mois, puis tous les 6 mois pendant 1 an, puis tous les ans jusqu’à l’âge de la puberté.
Ce protocole de surveillance permet d’améliorer les connaissances sur l’évolution des gliomes et de valider la prise en charge des jeunes enfants atteints de NF1.
I – PRISE EN CHARGE THÉRAPEUTIQUE :
1- Traitement des manifestations cutanées :
* Taches café au lait :
Des succès thérapeutiques ont été rapportées avec des lasers YAG, Q-switched ruby et pulsés sur les TCL.
Cependant, les résultats semblent temporaires. Des aggravations ont été rapportées.
Ce traitement ne peut donc être recommandé actuellement.
* Traitement des neurofibromes :
+ Neurofibromes cutanés :
Plusieurs méthodes peuvent être utilisées pour l’exérèse complète ou partielle de neurofibromes volumineux ou gênants.
Quand les neurofibromes cutanés sont de petite taille, moins de 2 cm, et nombreux, la méthode de destruction de choix est le laser CO2.
L’électrocoagulation peut être choisie quand les éléments sont peu nombreux.
La chirurgie classique est requise quand les lésions font plus de 2 cm.
Selon leur importance et/ou leur nombre, ces destructions peuvent être réalisées sous anesthésie locale ou générale.
Le laser CO2 sous anesthésie générale permet de détruire plusieurs centaines de neurofibromes en une seule séance.
+ Neurofibromes nodulaires périphériques :
La chirurgie d’exérèse des neurofibromes périphériques doit se faire sous microscope pour éviter des séquelles fonctionnelles neurologiques.
+ Neurofibromes plexiformes :
Le traitement ne peut être que chirurgical.
L’exérèse est le plus souvent intralésionnelle.
Les complications hémorragiques sont fréquentes.
L’autotransfusion doit être proposée. Les neurofibromes plexiformes de la face posent des problèmes complexes de réparation.
Il est aujourd’hui assuré que les destructions de neurofibromes n’entraînent aucun risque d’accélération de croissance des neurofibromes restants, ni de cancérisation.
Le bénéfice psychologique de telles destructions est souvent considérable, pouvant permettre d’espérer le maintien ou la reconquête d’une vie affective, sociale, psychologique, normale ou quasi normale.
2- Traitement des complications :
Certaines scolioses dystrophiques nécessitent un traitement chirurgical dans l’enfance ou à l’adolescence.
Le traitement des gliomes optiques est rarement justifié du fait de leur faible évolutivité.
L’indication thérapeutique (chimiothérapie, radiothérapie, chirurgie) est pesée suivant la localisation, l’évolution, le retentissement et l’âge.
Les traitements de TMGN fait appel à une chirurgie d’exérèse complète associée à une radiothérapie plus ou moins une chimiothérapie adjuvante par anthracyclines.
Neurofibromatose de type 2 (ancienne neurofibromatose acoustique ou centrale) :
A – ÉPIDÉMIOLOGIE ET GÉNÉTIQUE :
La NF2 était anciennement dénommée neurofibromatose acoustique.
Elle est beaucoup plus rare que la NF1, avec une incidence d’une naissance sur 33 000 à 40 000.
Elle est caractérisée par des schwannomes vestibulaires bilatéraux (anciennement neurinomes de l’acoustique), des schwannomes d’autres nerfs crâniens et spinaux, et des méningiomes.
Le gène de la NF2 a été identifié sur le chromosome 22 dans la région 22q12.2.
Ce gène est un gène suppresseur de tumeur.
La protéine produite a été appelée merline ; elle fait partie d’un groupe de protéines liées au cytosquelette.
La pénétrance du gène NF2 est complète à l’âge de 60 ans et les mutations de novo représentent environ 50 % des cas.
B – MANIFESTATIONS CLINIQUES :
Les manifestations cutanées sont inconstantes et le plus souvent discrètes.
Les TCL sont présentes dans environ la moitié des cas, beaucoup moins nombreuses qu’au cours de la NF1, généralement au nombre de deux.
Il n’y a pas de lentigines des plis dans la NF2.
Les tumeurs cutanées, schwannomes et moins fréquemment neurofibromes, sont présentes chez environ 70 % des individus affectés.
Elles sont peu nombreuses, moins d’une dizaine.
Ces schwannomes ont l’aspect, soit de tumeurs sous-cutanées sensibles à la pression, impossibles à distinguer cliniquement des neurofibromes nodulaires sous-cutanés, soit, plus fréquemment, de zones d’épaississement cutané en « plateau » à peine surélevé, pigmentées, souvent pileuses.
Il n’y a pas de neurofibromes plexiformes. Les anomalies oculaires sont fréquentes.
Soixante-dix pour cent des malades ont une cataracte juvénile postérieure.
Beaucoup plus rarement sont détectés des hamartomes rétiniens.
Les nodules de Lisch sont absents.
Les schwannomes vestibulaires sont quasiment constants (90 % des cas).
Le diagnostic en est généralement fait à la troisième décennie.
Des méningiomes sont présents dans 50 % des cas.
Des schwannomes du système nerveux central, d’autre localisation que vestibulaires, des épendymomes ou des neurofibromes spinaux sont fréquents. Les gliomes des voies optiques ne se retrouvent pas au cours de la NF2.
C – PRONOSTIC :
Le pronostic de la NF2 est catastrophique, avec une espérance de vie de l’ordre de 50 ans.
Trois phénotypes de gravité faible, moyenne ou sévère semblent se dégager.
La sévérité pronostique apparaît corrélée à la présence d’harmartomes rétiniens et de certaines mutations.
Le phénotype de gravité se retrouve chez les membres d’une même famille.
Le traitement des schwannomes vestibulaires dépend du retentissement fonctionnel, du nombre et de la taille des tumeurs et de l’âge du patient.
Son but principal est de préserver la fonction auditive.
Si la tumeur est à croissance lente et trop grande pour être excisée sans induire de surdité, résection partielle ou abstentionsurveillance sont indiquées.
Le traitement des autres tumeurs est le même que le patient ait ou non une NF2.
La prise en charge est assurée au mieux par des centres spécialisés pluridisciplinaires où oto-rhino-laryngologistes, neurologues et neurochirurgiens entraînés à ce type de pathologie associent leur compétence.
Neurofibromatose segmentaire (NF5) :
Les neurofibromatoses segmentaires sont exceptionnelles, avec une prévalence de moins de 0,001 %.
Elles sont caractérisées le plus souvent par la présence de neurofibromes, plus rarement de TCL, et parfois de lentigines ou exceptionnellement de nodules de Lisch sur un seul segment corporel, voire sur un hémicorps, ou plus rarement sur plusieurs segments bilatéraux.
Le diagnostic ne peut être porté qu’après avoir éliminé le diagnostic de NF1 ou de NF2.
Les neurofibromatoses segmentaires pourraient être une forme mosaïque de NF1.
Le conseil génétique doit être prudent car il existe quelques cas exceptionnels de NF1 héritée de parents ayant une forme segmentaire (impliquant alors des cellules germinales).
Formes rares de neurofibromatose :
A – NEUROFIBROMATOSE 3 OU NEUROFIBROMATOSE MIXTE :
Quelques cas exceptionnels de NF tiennent à la fois de la NF1 et de la NF2.
B – TACHES CAFÉ AU LAIT ISOLÉES OU NEUROFIBROMATOSE 6 :
Des TCL qui demeurent isolées à l’âge adulte dans au moins deux générations, parfois associées à d’autres anomalies de la pigmentation, définissent cette forme particulière de NF.
Des lentigines des plis, rarement des nodules de Lisch, sont parfois retrouvés, mais sans aucun neurofibrome.
Il y a parfois des anomalies osseuses (pectus excavatum, genu valgum, pieds plats), voire des difficultés scolaires non spécifiques.
La transmission est autosomique dominante.
Ces formes exceptionnelles peuvent être difficiles à distinguer du syndrome des lentigines multiples.
Il s’agirait de forme allélique de NF1.
C – NEUROFIBROMATOSE À DÉBUT TARDIF OU NEUROFIBROMATOSE 7 :
Il s’agit de NF avec neurofibromes d’apparition tardive après 30 ans.
D – SCHWANNOMATOSES OU NEURILEMMOMATOSES :
Les schwannomatoses sont caractérisées par des schwannomes multiples cutanés et sous-cutanés sans schwannomes vestibulaires.
Il s’agit donc d’une maladie proche de la NF2, mais de meilleur pronostic.
Conclusion :
Les NF constituent donc un ensemble hétérogène. Le classement nosologique de chaque cas est indispensable au suivi.
Selon les NF, le pronostic, les complications, le conseil génétique et la prise en charge thérapeutique sont différents.
Les centres multidisciplinaires de référence, avec leurs réseaux d’experts, sont les structures de choix d’accueil des malades.







")



{kind=link}