



















La maladie de Parkinson est une maladie fréquente après 60 ans, liée à une perte des neurones dopaminergiques dans la substance noire. Le diagnostic uniquement clinique repose sur l’asymétrie des signes, la rigidité, l’akinésie et l’amélioration prolongée par le traitement. Dans ces conditions, le risque d’erreur de diagnostic est inférieur à 20 %. La stratégie thérapeutique doit tenir compte de l’âge du patient, de son handicap et des signes associés tels des troubles psychiques. Très schématiquement, chez le sujet jeune, la réponse thérapeutique est excellente mais ces sujets vont relativement rapidement présenter des fluctuations d’effets thérapeutiques et des dyskinésies. Un traitement privilégiant les agonistes dopaminergiques ou l’association si nécessaire de doses modérées de L-dopa. À l’inverse, des patients plus âgés doivent bénéficier d’un traitement simple, le plus souvent en monothérapie par la L-dopa.
RAPPEL HISTORIQUE :
 En 1817, paraît l’ouvrage “ Assay on a shaking palsy ”, dans lequel sir James Parkinson fait une description clinique très précise de la “ paralysie agitante ”. Le terme “ akinésie ” est défini plus tard (par Wilson, en 1929).
En 1817, paraît l’ouvrage “ Assay on a shaking palsy ”, dans lequel sir James Parkinson fait une description clinique très précise de la “ paralysie agitante ”. Le terme “ akinésie ” est défini plus tard (par Wilson, en 1929).
En 1880, J.-M. Charcot traite ses patients avec de la scopolamine (anticholinergique), du fait de la séborrhée du visage qu’il avait attribuée à une hypercholinergie.
En 1913, Guggenheim, en Suisse, isole la dopa à partir de haricots (de Windsor). Il en donne quelques milligrammes à son chien qui vomit et il en conclut : “ peu d’intérêt ”.
En 1917-1925, l’épidémie d’encéphalite léthargique de von Economo fait plusieurs millions de morts. Elle a laissé parfois comme séquelle une maladie de Parkinson “ postencéphalitique ” qui a la particularité d’être peu évolutive (sur plus de 20 ans).
En 1948, le trihexyphénidyle (Artane), premier anticholinergique de synthèse, est commercialisé.
En 1951, P. Deniker montre l’intérêt de la chlorpromazine dans les troubles psychiatriques. Comme premier neuroleptique, on retrouve le Largactil, de “ large action ”, car efficace sur de nombreux symptômes psychiatriques.
En 1954, Carlsson, en Suède, découvre le mécanisme d’action des neuroleptiques : ils bloquent les récepteurs dopaminergiques.
En 1960-1965, l’existence d’un déficit en dopamine dans le cerveau des patients parkinsoniens est mise en évidence (Barbeau, Hornykiewicz et Birkmayer).
En 1965, on effectue les premiers essais avec la dopa (mélange racémique), puis avec la L-dopa. premiers succès, Cotzias (Etats-Unis).
En 1969, la L-dopa est commercialisée en France (Larodopa).
En 1974, La L-dopa est associée à un inhibiteur de la décarboxylase périphérique (bensérazide [Modopar ], carbidopa, [Sinemet ]).
En 1975, agonistes dopaminergiques (bromocriptine [Parlodel ], piribédil [Trivastal ]).
En 1982, la première greffe intracérébrale de glande surrénale est effectuée par Borklund en Suède.
En 1989, les premières greffes de cellules fœtales humaines dans le striatum sont réalisées.
En 1990, on découvre qu’un IMAO B (sélégiline [Déprényl ]) a un effet favorable possible sur la progression de la maladie (étude DATATOP sur 800 patients, Etats-Unis). En 1996, l’effet n’est pas confirmé.
En 1992, on assiste au début des stimulations chroniques intracérébrales (thalamiques) instaurées par A.-L. Benabid.
L-DOPA
La L-dopa, précurseur de la dopamine, franchit la barrière hémato-encéphalique. Apporté par voie sanguine, cet acide aminé ne présente par lui-même aucune propriété pharmacologique.
La L-dopa exerce son action centrale et périphérique par sa transformation en dopamine grâce à la dopa-décarboxylase, enzyme présente dans de nombreux tissus.
L’action de la L-dopa sur l’organisme est diffuse, centrale et périphérique : l’action sur la voie dopaminergique nigrostriée améliore les signes de la maladie, les autres actions sont responsables des effets indésirables (nausées, hypotension).
Pharmacocinétique :
– Les taux sanguins de L-dopa varient très vite du fait d’un métabolisme rapide qui s’effectue selon une décarboxylation en dopamine (70 %), une méthylation en 3-O-méthyl-dopa (3-OMD [10 %]).
– Sa demi-vie est courte (l heure) et son absorption rapide obéit à de grandes variations interindividuelles. Le pic plasmatique est atteint en 20 à 90 minutes. Les taux efficaces sont compris entre 1 000 et 3 000 ng/ml.
– L’heure de la prise, le repas, le régime alimentaire, les anticholinergiques et les pansements gastriques sont autant de facteurs de variations de l’absorption digestive.
– La relation entre la L-dopa ingérée et le pic plasmatique (C. max) n’est ni étroite ni linéaire. les faibles doses sont proportionnellement mieux absorbées.
– Au niveau gastrique, l’absorption est faible et la décarboxylation importante. Le rôle majeur de l’estomac est celui d’une valve régulatrice délivrant plus ou moins vite la L-dopa dans le jéjunum où elle est absorbée. Ainsi une prise unique peut provoquer deux pics plasmatiques, et l’absorption est très rapide chez les patients gastrectomisés. Maximale au niveau du jéjunum, elle peut être gênée par les acides aminés aromatiques (L-tryptophane, tyrosine, 3-O-méthyl-dopa).
– Pour pénétrer dans les tissus cérébraux, la L-dopa doit franchir deux barrières, l’une digestive et l’autre hémato-encéphalique. Ces traversées membranaires s’effectuent par un mécanisme de transport actif et saturable.
L-dopa tôt ou tard ?
– Apparue en 1970, la L-dopa n’est pas restée longtemps seule. Quatre ans plus tard, elle est associée à un inhibiteur de la décarboxylase périphérique qui permet de diviser les doses quotidiennes par cinq et d’ atténuer considérablement les effets secondaires de type périphérique (nausées et hypotension orthostatique).
– Dans les premières années d’utilisation, tous les patients parkinsoniens ont été traités par la L-dopa et souvent à des doses élevées, faisant apparaître des mouvements involontaires de surdosage et dévoilant des fluctuations d’efficacité du traitement au cours de la journée. De ce fait, après quelques années d’utilisation, des détracteurs, tout en reconnaissant l’efficacité souvent spectaculaire du médicament, l’accusaient de provoquer des mouvements anormaux et des fluctuations d’efficacité du traitement.
– Ainsi sont nées schématiquement deux attitudes : pour les partisans de la L-dopa, il fallait l’utiliser le plus tard possible pour tenter de différer les complications évolutives. pour les partisans de la dopathérapie, il faut l’utiliser le plus tôt possible pour utiliser le traitement au moment où il est le plus efficace. La divergence existe encore de nos jours, mais elle s’est très atténuée et on peut considérer maintenant qu’il faut donner la dopa “ ni trop tôt, ni trop tard ”.
L-dopa seule ou en association ?
La tendance est de donner la L-dopa assez rapidement, associée à une dose faible d’agoniste dopaminergique : c’est l’association précoce.
Cette façon de faire permet une action simultanée présynaptique et postsynaptique.
Elle a des avantages, dans l’immédiat, utilisant la synergie entre les deux produits et permettant peut-être, à long terme, de repousser l’apparition de fluctuations d’efficacité et de mouvements involontaires.
Quelle L-dopa prescrire ?
La dopathérapie s’est enrichie de deux nouvelles formes : les formes à libération prolongée (LP) et une forme buvable (dispersible). Schématiquement, ces trois formes se définissent par leur vitesse d’absorption.
On dispose ainsi de trois formes :
– à absorption rapide : la forme buvable.
– à absorption à vitesse intermédiaire : la forme standard.
– et à absorption plus lente et prolongée : la forme à libération prolongée.
Ces trois formes ont des indications précises, schématiquement :
– la plupart des patients sont redevables de la forme standard.
– les patients qui présentent des fluctuations d’efficacité de type sont redevables de la forme à libération prolongée.
– et la forme buvable peut être utilisée ponctuellement lorsqu’il existe un besoin rapide de L-dopa dans la journée ou encore chez les patients qui présentent des troubles de la déglutition, indication pour laquelle elle a été mise au point initialement.
PHYSIOPATHOLOGIE :
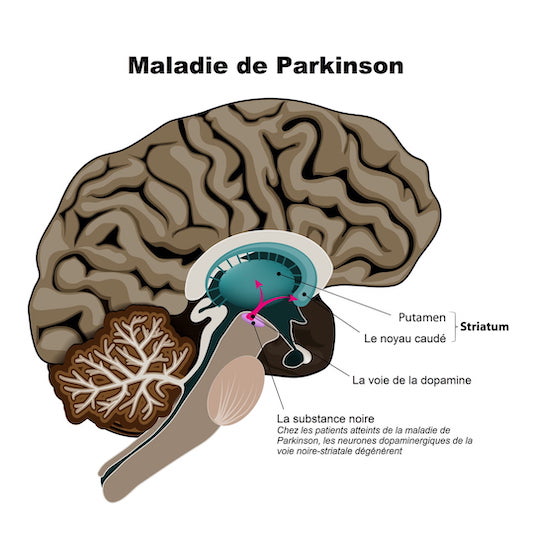
La lésion responsable de la maladie de Parkinson siège dans le locus niger, noyau de quelques millimètres de long, en forme de croissant, situé dans la partie haute du tronc cérébral.
Cette structure comprend les corps cellulaires des neurones dopaminergiques contenant de la mélanine. Elle est pigmentée en noir, d’où son nom.
Ce groupe de neurones transmet l’influx nerveux grâce à un neuromédiateur : la dopamine.
Voie nigrostriée :
La voie nigrostriée est constituée des prolongements des neurones dopaminergiques dont les corps cellulaires sont situés dans la pars compacta du locus niger.
Leurs axones se distribuent au striatum (noyau caudé et putamen) avec quelques collatérales à destination pallidale.
Cette voie dopaminergique exerce un rôle inhibiteur sur l’activité des neurones cholinergiques du striatum.
La baisse de concentration en dopamine, dans le striatum, provoque une hypercholinergie.
La thérapeutique consiste soit à prescrire des anticholinergiques, soit à restaurer le fonctionnement des neurones dopaminergiques par la L-dopa ou des agonistes dopaminergiques directs.
On admet que les premiers signes cliniques apparaissent lorsqu’une baisse de 80 % de la dopamine est atteinte dans le striatum.
Autres lésions :
Il existe d’autres systèmes touchés dans la maladie de Parkinson : le locus cœruleus (régulation de la tension artérielle et de la fréquence cardiaque), le noyau dorsal du vague (motricité digestive, vésico-sphinctérienne, salivation), l’hypothalamus (température, poids corporel), les ganglions sympathiques périphériques…
Dans toutes ces structures où siège une perte neuronale modérée sans rapport avec l’importance de l’atteinte du locus niger, une diminution des concentrations en dopamine, noradrénaline et sérotonine est détectée.
Corps de Lewy :
L’étude en microscopie permet de retrouver la présence de corps de Lewy, inclusions intracytoplasmiques situées dans les neurones lésés qui affirment le diagnostic de maladie de Parkinson idiopathique (ils sont, par exemple, absents dans la paralysie supranucléaire progressive ou maladie de Steele-Richardson).
La nature de ces inclusions est inconnue.
DIAGNOSTICS DIFFERENTIELS :
Maladie de Parkinson idiopathique :
La maladie de Parkinson idiopathique (MPI) représente la cause la plus fréquente des syndromes parkinsoniens.
Cette maladie dégénérative, dont l’origine est inconnue, débute à l’âge moyen de 60 ans (de 30 à 85 ans). Avant 40 ans, il s’agit de maladie dite “ à début précoce ” :
– sur le plan anatomique, elle se caractérise par une dépopulation neuronale du locus niger (substance noire) prédominant dans la pars compacta, associée à une réaction gliale et à la présence de corps de Lewy.
– sur le plan biochimique, le fait essentiel réside dans la diminution de la concentration en dopamine dans le striatum (putamen et noyau caudé). La dégénérescence de la voie dopaminergique nigrostriée est corrélée à la sévérité des signes moteurs.
– le diagnostic clinique est facile, dans la majorité des cas, devant un tremblement de repos unilatéral ou une micrographie. La réponse thérapeutique à la L-dopa en est une bonne confirmation, si bien qu’il est rarement utile de demander des examens complémentaires. Mais la moindre “ aspérité ”, tant à l’interrogatoire qu’à l’examen clinique, doit conduire à rechercher une autre étiologie.
Autres syndromes parkinsoniens :
Dans les autres syndromes parkinsoniens, on retrouve :
– en premier lieu, un syndrome parkinsonien iatrogène dû aux neuroleptiques car il est réversible.
– les autres diagnostics sont essentiellement représentés par les “ faux parkinsons ” : paralysie supranucléaire progressive, atrophies multi-systématisées, dégénérescence cortico-basale et maladies des corps de Lewy, chiffrées à 10-20 % de la totalité des syndromes parkinsoniens. Ils sont souvent difficiles, au début, parfois établis après plusieurs années de suivi ou même post-mortem.
– ensuite, il s’agit de situations exceptionnelles : maladie de Wilson chez le sujet jeune, maladie de Fahr.
– une autre circonstance de diagnostic subtil est la survenue de chutes ou de troubles de la marche isolés chez un sujet âgé : hydrocéphalie à pression normale, état lacunaire, maladie de Parkinson idiopathique ou… conséquence du grand âge.
NOUVEAUX TRAITEMENTS :
Agonistes dopaminergiques :
Les récepteurs dopaminergiques postsynaptiques sont peu atteints dans la maladie de Parkinson et la recherche de nouveaux composés continue pour des agents plus spécifiques, plus efficaces et ayant moins d’effets secondaires.
Après la bromocriptine, le piribédil, le lisuride, le ropinirole, d’autres agonistes sont en cours de développement.
Actuellement, aucun de ces composés n’a un rapport efficacité-tolérance aussi bon que celui de la L-dopa, mais ils constituent un apport indispensable.
Inhibiteurs de la catéchol-O-méthyl-transférase :
Les inhibiteurs de la catéchol-O-méthyl-transférase (ICOMT), en modifiant la cinétique plasmatique de la lévodopa, vont avoir un effet bénéfique sur les fluctuations d’efficacité liées aux prises de L-dopa.
Stimulation intracérébrale :
La stimulation intracérébrale à haute fréquence par électrodes implantées à demeure est utilisée, depuis 1992, pour traiter les cas de tremblement résistant au traitement.
Auparavant, dans les années 60, on pratiquait une lésion définitive par échauffement (thalamotomie, pallidotomie).
L’électrode implantée est alimentée par un boîtier sous-cutané (identique à celui d’un stimulateur cardiaque), mis en route par le patient lui-même ou par un programmateur.
Les meilleures indications sont un tremblement unilatéral bien que des stimulations bilatérales soient possibles.
De la même manière, la stimulation pallidale permet de réduire considérablement les mouvements anormaux d’un hémicorps induits par la L-dopa.
La stimulation bilatérale des deux noyaux sous-thalamiques qui permet d’améliorer l’état des patients “ on-off ” est en cours d’évaluation.
Implantations de tissus dopaminergiques intracérébrales :
Les greffes de tissus dopaminergiques dans le noyau caudé ont été réalisées, dès 1981, par des médecins suédois. D’autres expériences ont été conduites avec plus ou moins de succès.
Au début, il s’agissait d’implanter du tissu issu de la médulo-surrénale du patient lui-même. La médulo-surrénale possède des équipements enzymatiques pour synthétiser les catécholamines, en particulier la noradrénaline et la dopamine.
Plus tard, les chercheurs ont implanté du tissu dopaminergique d’un embryon humain, âgé de 8 semaines, dans le noyau caudé. Cette pratique est beaucoup plus simple sur le plan chirurgical.
Les résultats obtenus demeurent néanmoins inconstants et restent dans le domaine expérimental.
INTERACTIONS MEDICAMENTEUSES :
Neuroleptiques
Les phénothiazines et les butyrophénones, mais aussi les autres agents pharmacologiques bloquant les récepteurs dopaminergiques, benzamides substituées (métoclopramide), s’opposent à l’action de la L-dopa sur le récepteur postsynaptique.
Il est donc illogique de les associer à la L-dopa. Seule la dompéridone, neuroleptique dont l’action est uniquement périphérique, est possible.
Antihypertenseurs
Les antihypertenseurs doivent être utilisés avec discernement et prudence.
On évitera les antihypertenseurs centraux (alpha-méthyl-dopa, clonidine), car ils peuvent interférer avec le métabolisme de la dopamine.
On leur préférera, plutôt que les diurétiques, les bêta-bloquants, les inhibiteurs des canaux calciques ou de l’enzyme de conversion.
Antidépresseurs
Les antidépresseurs s’avèrent souvent nécessaires.
Les tricycliques ont un effet anticholinergique favorable, mais un effet alpha-bloquant défavorable et ils peuvent majorer une hypotension orthostatique.
Si tel est le cas, l’emploi de molécules non tricycliques est préférable. On préférera des molécules sédatives prescrites le soir à faible dose (miansérine [Athymil ]). La fluoxétine (Prozac) est contre-indiquée avec la sélégiline (Déprényl).
Amines sympathomimétiques
Les amines sympathomimétiques (adrénaline, isoprénaline, éphédrine, dobutamine), rarement indiquées, doivent être évitées car leurs effets sur les récepteurs adrénergiques périphériques s’ajoutent à ceux de la dopamine (variations tensionnelles).
Inhibiteurs de la monoamine oxydase
Les inhibiteurs de la monoamine oxydase (IMAO) non sélectifs sont déconseillés car, plus qu’ailleurs, ils risquent de provoquer des variations brutales et incontrôlables de la pression artérielle.
Cependant, les IMAO de type B, comme la sélégiline (Déprényl), qui agissent spécifiquement sur la dopamine, sont proposés comme traitement adjuvant de la dopathérapie.
Vitamine B6
La vitamine B6, cofacteur de la décarboxylase périphérique, accélère la dégradation de la L-dopa en dopamine.
Si la L-dopa est utilisée seule (Larodopa), ce qui est maintenant exceptionnel, la vitamine B6 est contre-indiquée.
En revanche, si elle est associée à un inhibiteur de la décarboxylase, la vitamine B6 reste logiquement sans effet néfaste.
Agents cholinoformateurs
Les agents cholinoformateurs (déanol, diméthyl-amino-éthanol) peuvent avoir une action contraire à celle de la L-dopa par leur effet stimulant sur l’activité des neurones cholinergiques centraux (striatum).
Papavérine
La papavérine est théoriquement déconseillée car elle s’oppose expérimentalement à l’action de la dopamine.
En fait, aucun essai clinique, chez des patients traités, ne démontre cet antagonisme. Cette contre-indication est donc toute relative.
CHUTES :
Les chutes s’observent après plusieurs années d’évolution sauf chez le sujet âgé.
Elles peuvent avoir plusieurs mécanismes.
Chutes accidentelles
Les chutes sont accidentelles lorsque le patient a trébuché sur un obstacle. elles sont facilitées par la maladie de Parkinson.
Chutes par “ freezing ”
Tous les stimuli visuels ou auditifs peuvent entraîner un blocage brutal de la motricité responsable d’un déséquilibre et d’une chute : le passage d’une porte, une émotion ou un événement inattendu mais aussi familier comme la sonnerie du téléphone.
La chute est généralement précédée d’un piétinement, les pieds sont comme “ collés au sol ” et le patient s’incline vers l’avant, d’un bloc, “ en statue ” ou pivote sur le côté.
Chutes par instabilité permanente
Les chutes par instabilité permanente se voient chez les patients ayant plusieurs années d’évolution, lorsque s’est installée une instabilité à la marche, sans vertiges, non latéralisée, sans élargissement du polygone de sustentation et sans déficit proprioceptif.
Cette instabilité, qui, sur le plan physiopathologique, n’est pas élucidée, peut provoquer une chute, surtout à l’occasion d’un changement de direction, d’un ralentissement ou à l’arrêt.
Ces chutes se font parfois en arrière et peuvent être graves.
Chutes liées à une hypotension orthostatique
Les chutes sont liées à une hypotension orthostatique lorsque le patient change de position brutalement, en se levant brusquement d’un lit ou d’un fauteuil.
Le diagnostic est relativement aisé par l’interrogatoire du patient qui précise que la chute a été précédée d’un malaise.
La prise de la tension artérielle couché puis debout à plusieurs reprises pourra confirmer l’hypotension orthostatique à pouls variable ou à pouls constant traduisant alors une dysautonomie (ou la présence d’un traitement par bêta-bloquant).
Diagnostic positif
Nous prendrons pour type de description la maladie de Parkinson idiopathique (MPI).
Le diagnostic de la maladie de Parkinson est avant tout clinique, aussi s’assurera-t-on, tant par un interrogatoire que par un examen clinique, qu’aucune autre étiologie n’est responsable de ce syndrome extra-pyramidal. Au besoin, on s’aidera de quelques examens complémentaires.
Les signes de début sont souvent unilatéraux ou asymétriques avec apparition :
– d’un tremblement de repos de la main ou du pied.
– d’une écriture qui devient plus petite (micrographie).
– d’un ralentissement global de l’activité (lenteur).
– de troubles de la marche.
– d’une sensation de raideur avec gêne à l’exécution de certains gestes comme celui de mettre un manteau.
– d’une fatigabilité anormale simulant un syndrome dépressif.
– de douleurs diffuses qu’elles soient musculaires ou articulaires (PASH).
Le tableau clinique s’installe progressivement, en quelques mois, et se complète.
On distingue ainsi les formes akinéto-hypertoniques (30 % des cas), les formes complètes et les formes tremblantes habituellement moins évolutives.
Les signes restent très longtemps asymétriques.
SIGNES NEUROLOGIQUES :
Tremblement de repos :
Le tremblement parkinsonien est un tremblement de repos. Il disparaît aussi bien au cours de l’exécution du mouvement que lors du maintien d’une attitude et réapparaît dès l’arrêt du mouvement.
Cependant, lorsque le tremblement est important, il peut persister lors du mouvement mais de manière atténuée.
– Le tremblement :
– est lent, 4 à 7 cycles par seconde, et régulier.
– prédomine aux extrémités.
– est aggravé par les émotions et disparaît pendant le sommeil.
– augmente ou il est déclenché par l’ épreuve du calcul mental, ce qui le différencie des autres tremblements.
– On le compare à des mouvements comme celui d’émietter du pain, de rouler une cigarette. Les avant-bras “ battent le tambour ”, le pied effectue des petits mouvements de pédalage. Le menton ou la lèvre inférieure peuvent être concernés mais il épargne toujours le chef.
– Chez quelques patients, on peut observer la coexistence d’un tremblement d’attitude (rapide, irrégulier) et d’un tremblement de repos (plus lent).
– Il manque souvent dans les autres syndromes parkinsoniens qui se présentent comme des formes akinéto-hypertoniques pures.
Akinésie :
Perte de l’initiation et de l’exécution
L’akinésie est la perte de l’initiation et de l’exécution automatique du mouvement :
– le geste est lent, rare, réduit en amplitude et demande au malade un effort tout au long de son déroulement.
– le démarrage du pas se fait avec hésitation ou retard ( piétinement au démarrage).
– le balancement des bras a disparu, de même que le clignement des paupières ou la mimique ( faciès figé ou “ poker face ”).
– la parole devient monotone, souvent plus basse (comme enrouée), parfois le débit s’accélère (tachyphémie).
– l’écriture se modifie (la micrographie est parfois le premier signe), non pas par le tremblement qui n’est présent qu’au repos, mais par l’akinésie qui la rétrécit progressivement de ligne en ligne.
Lenteur des gestes
L’ensemble des gestes se fait avec lenteur, aussi bien les mouvements des mains, comme se boutonner, en particulier les gestes alternatifs, comme brosser ou de rotation, comme battre des œufs, que les mouvements axiaux, comme se lever d’une chaise.
Chaque patient présente une gêne motrice qui lui est propre.
Epreuves simples
Des épreuves simples évaluent l’ akinésie segmentaire : “ faire des marionnettes ”, mouvements rapides d’ouverture et de fermeture de la pince pouce-index ou d’ouverture et de fermeture de la main, tapotement du pied sur le sol. on comparera le côté droit et le côté gauche.
L’ akinésie axiale se traduit dans les mouvements du tronc : se lever d’un siège, sortir d’une voiture ou se retourner dans le lit.
Disparition de l’akinésie axiale
Cette akinésie peut s’effacer totalement et soudainement, durant quelques instants, et le malade retrouve alors des possibilités normales (marche, parole, écriture) :
– ce sont les “ kinésies paradoxales ” souvent déclenchées par une vive émotion.
– elles caricaturent la variabilité des signes de la maladie de Parkinson d’un moment à l’autre, d’une journée à l’autre. Il s’agit d’une des particularités de cette affection.
Perte du mouvement
L’akinésie est le signe le plus difficile à décrire et à évaluer : il ne s’agit pas d’une paralysie, mais d’une “ perte du mouvement ”.
Ce signe majeur de la maladie est le plus handicapant, mais aussi le moins visible, contrairement au tremblement de repos très visible mais rarement source de handicap grave.
Au stade évolué, l’akinésie est responsable de troubles de la marche et de chutes.
Hypertonie extra-pyramidale :
– L’hypertonie extra-pyramidale est plastique, de consistance cireuse ou en “ tuyau de plomb ”, le membre examiné conservant l’attitude qui lui a été donnée.
– Elle reste stable et homogène tout au long de la mobilisation passive, contrairement à l’hypertonie pyramidale, dite “ spastique élastique ”, qui se renforce d’autant plus que l’étirement passif est rapide.
– Le phénomène de la “ roue dentée ” est perçu au niveau du tendon du biceps lors de l’extension lente de l’avant-bras sur le bras : l’hypertonie extra-pyramidale cède par à-coups, mais le sujet doit être parfaitement détendu :
– ce signe est inconstant et il arrive de rencontrer des faux positifs si le patient n’est pas détendu ou s’il existe un tremblement d’attitude associé.
– on retrouve également une augmentation du réflexe de posture, bien perceptible au niveau du tendon du jambier antérieur.
– La “ manœuvre de Froment ” accentue l’hypertonie extra-pyramidale : tout en effectuant des mouvements lents, passifs, de flexion-extension de l’avant-bras sur le bras, on demande au patient de “ faire les marionnettes ” avec l’autre main : cela a pour effet de renforcer (ou de faire apparaître) l’hypertonie du bras examiné.
– L’hypertonie prédomine sur les muscles antigravidiques du cou et du tronc et aux racines des membres :
– à un stade évolué, le malade prend une attitude caractéristique : la tête est projetée en avant, le cou en extension, en “ col de cerf ”, le dos est courbé, les bras demi-fléchis sont collés au corps et les genoux et les hanches sont en demi-flexion.
– les doigts en extension réalisent la “ main d’écrivain ” et les déformations du pied apparaissent plus tardivement, en équin avec flexion en griffe des orteils.
Autres signes neurologiques :
Le réflexe naso-palpébral est vif et inépuisable.
Les réflexes ostéo-tendineux sont normaux. Il n’y a pas de signe de Babinski, de signes cérébelleux, de déficit sensitif et l’oculomotricité est normale.
AUTRES SIGNES CLINIQUES :
Signes neuro-végétatifs :
Parmi les signes neuro-végétatifs, on retrouve :
– une hypotension orthostatique qui risque d’être majorée par le traitement.
– une hypersalivation, séborrhée du visage qui est luisant (“ visage pommadé ”), parfois larmoiement, fausse conjonctivite, dermite séborrhéique du cuir chevelu.
– des troubles vésico-sphinctériens (dysynergie vésico-sphinctérienne, contraction désinhibée du détrusor), responsables d’envies fréquentes (à différencier d’un obstacle prostatique chez l’homme : même âge).
– des troubles de la sudation.
Troubles psychiques :
Troubles de l’humeur
Des troubles de l’humeur sont fréquents et un état dépressif peut accompagner le début de la maladie ou apparaître plus tardivement.
Pour certains auteurs, la dépression serait “ le quatrième signe ” de la maladie.
Cet état dépressif est rarement intense, car les idées suicidaires et les sentiments d’auto-accusation sont peu fréquents. Il s’accompagne très souvent d’anxiété et pose à la fois des problèmes diagnostiques (lenteur de l’akinésie ou ralentissement de la dépression ?) et thérapeutiques (tolérance des antidépresseurs, interactions médicamenteuses).
Anxiété
L’anxiété est fréquente, tout au long de l’évolution, avec une hyperémotivité. Elle interfère avec certains signes moteurs (tremblement).
Hallucinations visuelles
Des hallucinations visuelles surviennent par accès de courte durée, généralement en fin de journée, après quelques années d’évolution, favorisées par les traitements.
Il s’agit de personnages que le patient connaît et reconnaît, parfois d’un animal domestique, de silhouettes ou d’hallucinations “ de présence ” : le patient croit à la présence de quelqu’un dans la pièce.
Ces hallucinations sont généralement critiquées, peu gênantes et n’annoncent pas forcément le début d’une détérioration intellectuelle.
Accès confusionnels
Des accès confusionnels surviennent chez les sujets âgés, d’origine médicamenteuse ou à l’occasion d’une déshydratation, d’un foyer infectieux (dont c’est parfois le premier signe).
Détérioration intellectuelle
On observe une détérioration intellectuelle de type sous-cortico-frontal avec ralentissement psychomoteur et difficultés de concentration, d’attention et d’apprentissage.
Signes généraux :
Dans les signes généraux, on retrouve :
– un amaigrissement (parfois important, de plusieurs kilogrammes, qui peut inaugurer la maladie).
– des œdèmes des membres inférieurs.
– il faut rechercher de principe une dysthyroïdie : si fixité du regard, tremblement, amaigrissement : hyperthyroïdie. à l’inverse si fatigue, lenteur : hypothyroïdie.
EXAMENS COMPLEMENTAIRES :
L’électroencéphalogramme, le scanner, l’IRM et l’étude du liquide céphalo-rachidien sont normaux dans la maladie de Parkinson.
Le moindre doute sur une autre étiologie d’un syndrome extra-pyramidal doit conduire à les prescrire.
Diagnostic différentiel
La maladie de Parkinson idiopathique représente la cause la plus fréquente des syndromes parkinsoniens.
Mais la moindre “ aspérité ”, tant à l’interrogatoire qu’à l’examen clinique, doit conduire à rechercher une autre étiologie.
Les autres syndromes parkinsoniens sont plus rares (15 % des syndromes parkinsoniens).
En premier lieu, un syndrome parkinsonien iatrogène dû aux neuroleptiques doit être recherché car il est réversible. Les autres diagnostics sont essentiellement représentés par les “ faux parkinsons ” : paralysie supranucléaire progressive, atrophies multisystématisées, dégénérescence cortico-basale et maladies des corps de Lewy.
Ces autres syndromes parkinsoniens regroupent des étiologies variées.
Syndrome parkinsonien postencéphalitique :
Le syndrome parkinsonien postencéphalitique a surtout un intérêt historique.
Après l’épidémie d’encéphalite léthargique de von Economo, de 1918 à 1927, de nombreux syndromes parkinsoniens sont apparus parmi les survivants.
Les lésions prédominaient sur le locus niger et le pallidum. Il n’existait pas de corps de Lewy, mais une dégénérescence neuro-fibrillaire.
L’évolution était très lente sur plus de 20 ans. Il n’y a actuellement pratiquement plus de survivants.
Parmi les autres encéphalites, on ne peut retenir que l’encéphalite japonaise B pour laquelle le syndrome parkinsonien apparaît bénin et non évolutif.
Syndrome parkinsonien iatrogène :
C’est le tout premier diagnostic à éliminer car il est aisément curable. Tous les neuroleptiques peuvent l’induire.
– Les neuroleptiques classiques sont les phénothiazines, les butyrophénones et les benzamides substituées. Le diagnostic peut être évident chez un patient dont le traitement et la pathologie psychiatrique sont connus.
– Le diagnostic peut être beaucoup plus difficile en cas de traitement prolongé :
– par le métoclopramide (Primpéran), le tiapride (Tiapridal) ou le sulpiride (Dogmatil) prescrits pour des troubles digestifs.
– ou la flunarizine (Sibélium) pour des vertiges.
– le Noctran et la Mépronizine pour une insomnie.
– ou le véralipride (Agréal) pour des bouffées de chaleur.
– Plus récemment, des syndromes extra-pyramidaux ont été décrits sous inhibiteurs calciques : diltiazem (Tildiem).
La survenue des troubles dépend peu de la dose totale administrée. Ils peuvent apparaître précocement ou, au contraire, s’installer progressivement après plusieurs mois ou plusieurs années de traitement.
Ils régressent à l’arrêt du traitement, en plusieurs mois. Exceptionnellement, ils dévoilent une authentique maladie de Parkinson idiopathique, qui, elle, va progresser.
Paralysie supranucléaire progressive :
La paralysie supranucléaire progressive (PSP), ou maladie de Steele-Richardson-Olszewski, se caractérise par des lésions de dégénérescence neuro-fibrillaire prédominant dans le locus niger, le striatum, le pallidum et les tubercules quadrijumeaux.
– Le syndrome parkinsonien de forme akinéto-hypertonique, symétrique se traduit par une raideur axiale, une dysarthrie et des chutes précoces.
– Il s’accompagne d’une paralysie des mouvements de poursuite oculaire vers le bas puis en latéralité et en convergence.
– Un syndrome pseudo-bulbaire et une démence sous-cortico-frontale s’installent rapidement.
– L’évolution est rapide (5 à 7 ans).
Atrophies multisystématisées :
Dégénérescence striatonigrique
Dans la dégénérescence striatonigrique, les lésions prédominent au niveau du putamen, des globes pâles, du locus niger et du locus cœruleus.
Aux signes extra-pyramidaux s’ajoutent des signes pyramidaux, des manifestations dystoniques, une incontinence urinaire et une hypotension orthostatique.
Atrophie olivo-ponto-cérébelleuse
L’atrophie olivo-ponto-cérébelleuse (forme sporadique) associe des lésions de l’olive bulbaire, des noyaux du pont et une atrophie corticale cérébelleuse.
Le syndrome parkinsonien peut être inaugural, mais il est souvent plus tardif, précédé par des troubles cérébelleux.
Syndrome de Shy et Drager
Dans le syndrome de Shy et Drager, les lésions sont situées dans le locus niger et la colonne intermédiolatéralis de la moelle.
Le syndrome parkinsonien akinéto-hypertonique est rapidement associé à une dysautonomie sévère, en particulier une hypotension orthostatique majeure (à pouls constant), responsable de chutes précoces par perte de connaissance brève.
Dégénérescence cortico-basale :
La dégénérescence cortico-basale est au début unilatérale : hypertonie d’un membre avec dystonie d’une main, apraxie unilatérale.
Ensuite apparaissent des troubles de la marche, un syndrome pseudo-bulbaire et une démence.
Maladie des corps de Lewy diffus :
La maladie des corps de Lewy diffus débute par des signes extra-pyramidaux.
Ils sont rapidement suivis par des troubles psychiques (confusion et hallucinations) :
– dont la particularité est d’être fluctuants au cours de la journée.
– la prise d’antiparkinsoniens et de psychotropes (benzodiazépines, antidépresseurs, neuroleptiques) aggrave ou déclenche les troubles psychiques.
– une détérioration intellectuelle s’installe rapidement.
– l’évolution des signes moteurs et des troubles psychiques est rapide : de 4 à 5 ans.
Sur le plan anatomique, on retrouve des corps de Lewy dans les noyaux gris centraux, mais aussi au niveau du cortex cérébral.
Autres diagnostics différentiels :
Maladie d’Alzheimer
Il n’est pas rare d’observer des signes extra-pyramidaux au cours d’une démence d’Alzheimer.
L’existence d’un syndrome aphaso-apraxo-agnosique, absent dans la démence parkinsonienne, permet de différencier ces deux maladies.
Le diagnostic différentiel est beaucoup plus difficile avec la maladie des corps de Lewy. Il est souvent neuropathologique.
Hydrocéphalie à pression normale
L’hydrocéphalie à pression normale avec sa triade d’Hakim (syndrome frontal, troubles de la marche, troubles urinaires) peut évoquer un syndrome extra-pyramidal.
La dilatation des ventricules cérébraux sans atrophie corticale au scanner ou à l’IRM et l’amélioration des signes après ponction lombaire évacuatrice permettent le diagnostic.
Etat lacunaire
L’état lacunaire doit être évoqué chez un patient âgé, lorsque le syndrome extra-pyramidal se manifeste, surtout par des troubles de la marche, et lorsqu’il existe des antécédents d’hypertension artérielle.
Le scanner cérébral, ou mieux l’ IRM, permettent de mettre en évidence des lacunes multiples dans les noyaux gris centraux, souvent associées à des lésions de leuco-encéphalopathie liée à l’hypertension artérielle.
Syndromes parkinsoniens associés
Les syndromes parkinsoniens sont associés à une démence et une sclérose latérale amyotrophique et sont observés dans certaines îles du Pacifique occidental (l’île de Guam).
Il existe une perte neuronale et une dégénérescence neurofibrillaire.
Maladie de Wilson
La maladie de Wilson est une affection génétique rare, liée à un trouble du métabolisme du cuivre, à laquelle il faut penser devant tout syndrome extra-pyramidal débutant chez un sujet jeune (avant 50 ans).
– Les troubles peuvent être précoces vers l’âge de 15 ans et débuter par des manifestations hépatiques (cirrhose), une rigidité extra-pyramidale, des mouvements anormaux, un tremblement oppositionniste, des troubles psychiques constants.
– A l’examen oculaire à la lampe à fente, on retrouve un anneau vert péricornéen de Keyser-Fleisher. le dosage de la cuprémie, cuprurie et de la céruloplasmine permettent le diagnostic.
– Un diagnostic précoce et un traitement approprié entraînent une amélioration des signes neurologiques, voire leur disparition, tout comme les lésions hépatiques.
Causes rares
Parmi les causes rares, on retrouve :
– la maladie de Hallervorden-Spatz où l’on observe des microcalcifications et une perte neuronale dans le locus niger et les noyaux gris.
– la maladie de Fahr, dont la symptomatologie clinique est variable, qui se caractérise par des calcifications visibles radiologiquement dans les noyaux gris.
– les syndromes parkinsoniens symptomatiques qui sont rares. Des traumatismes crâniens répétés, comme chez les boxeurs, peuvent conduire à un syndrome parkinsonien atypique secondaire à des lésions vasculaires.
– des lésions vasculaires des noyaux gris centraux discutées par certains auteurs.
– des tumeurs intraparenchymateuses, gliomes, métastases, tuberculomes… dans les noyaux gris ou le locus niger. ou extra-parenchymateuses, comme les méningiomes de la ligne médiane, qui sont exceptionnelles.
– l’intoxication aiguë par l’oxyde de carbone pouvant entraîner une nécrose des globes pâles. Le syndrome parkinsonien apparaît dans les semaines suivant l’intoxication, après la période de coma.
– le manganèse, lors d’une intoxication chronique chez les ouvriers manipulant le minerai, induit un syndrome parkinsonien associé à un syndrome pyramidal et un tremblement d’attitude important. Le plomb est exceptionnellement en cause.
– chez des héroïnomanes, l’action toxique du MPTP (méthyl-phényl-tétrahydropyridine) a été responsable de cas de syndrome parkinsonien sévère.
Évolution
La L-dopa et les antiparkinsoniens ont transformé radicalement le devenir des patients, leur permettant, au moins les premières années de traitement, de mener une vie presque normale.
Ces traitements substitutifs ne ralentissent pas la progression de l’affection. Le bénéfice, souvent spectaculaire, apporté au patient les premières années de traitement, diminue au cours du temps. La L-dopa a réduit la mortalité d’un facteur 3 à un facteur 1,5.
La progression de la maladie est très variable selon les patients :
– les formes où prédomine le tremblement évoluent plus lentement.
– l’âge de début influe plus sur la durée d’évolution que sur la vitesse évolutive.
– les patients réagissant bien à la L-dopa, la première année de traitement, ont un bon pronostic.
– si un épisode confusionnel survient la première année de traitement, le pronostic intellectuel est mauvais.
– les patients chez qui la maladie débute tôt, avant 40 ans, ont un risque plus élevé de voir apparaître des fluctuations d’efficacité et des mouvements anormaux que les patients chez qui la maladie débute tard. Ces derniers sont exposés au risque de perte d’efficacité du traitement, de chutes, de troubles psychiques.
Schématiquement, il est possible de distinguer trois étapes évolutives.
Début de la maladie :
Au début de la maladie, le patient est peu gêné (stades 1 et 2 de Hoehn et Yahr).
Cette période appelée “ lune de miel ” dure en moyenne 5 ans.
La persistance de quelques signes (tremblement intermittent, perte du ballant d’un bras, micrographie, dysarthrie) n’empêche pas le patient de mener une vie pratiquement normale.
Lorsque la maladie est installée :
Lorsque la maladie est installée commence la période des complications évolutives :
– la gêne motrice est alors nette (stade 3 et 5 de Hoehn et Yahr). L’activité professionnelle se révèle soit difficile, soit impossible. Le handicap moteur amène le patient à restreindre sa vie familiale et sociale, les déplacements à l’extérieur sont limités.
– plusieurs degrés de handicap existent à ce stade, dont l’appréciation est gênée par des mouvements anormaux ou des fluctuations d’efficacité du traitement dans la journée : à certains moments, le patient est totalement libre de ses mouvements (période “ on ”, stade 2), à d’autres, il peut être très akinétique (période “ off ”, stade 5).
– ce stade évolue sur plusieurs années, l’apparition de chutes ou d’épisodes confusionnels annonce souvent le troisième stade évolutif.
Déclin d’efficacité :
Lors du déclin d’efficacité, le handicap est important (stade 4 à 5 de Hoehn et Yahr).
– Les chutes apparaissent et la marche devient impossible ou très difficile nécessitant une aide. Les gestes sont lents, rares, réduits au minimum :
– la parole, très atteinte, rend toute communication difficile. Une aide devient indispensable pour tous les actes de la vie quotidienne.
– des déformations articulaires fréquentes touchent les extrémités ou le tronc.
– à ce stade, des troubles psychiques peuvent exister : hallucinations visuelles, accès confusionnels ou détérioration intellectuelle.
– c’est le stade de la perte d’efficacité de la L-dopa. Il s’agit de patients chez qui la maladie évolue depuis de nombreuses années (plus de 10 ans) et souvent âgés.
Complications sous traitement
Schématiquement, quatre grandes complications évolutives peuvent survenir après quelques années de suivi et représentent un important facteur de handicap.
Les fluctuations et les mouvements anormaux concernent plutôt les sujets chez qui la maladie a débuté tôt (avant 50 ans), les troubles psychiques et la perte d’efficacité se rencontrant chez les sujets pour qui la maladie a débuté tard (après 70 ans) ou chez les sujets âgés.
FLUCTUATIONS MOTRICES :
Les fluctuations motrices sont de plusieurs types.
Fluctuations liées aux prises de la L-dopa :
Akinésies de “ fin de dose ”
Les akinésies de “ fin de dose ” sont les plus fréquentes :
– il s’agit d’une diminution progressive de la durée de l’effet de la prise de L-dopa qui donnait jusqu’alors une bonne efficacité sur plusieurs heures.
– il ne s’agit pas d’une perte d’efficacité car l’effet, bien qu’écourté, reste de bonne qualité, mais d’un épuisement précoce.
– l’akinésie réapparaît 2 à 3 heures après chaque prise, selon les patients en fin de matinée, en fin d’après-midi ou de soirée.
– les fluctuations sont de gravité variable :
– elles sont peu intenses, se manifestant par un simple ralentissement des gestes, n’entravant que peu l’autonomie.
– parfois elles sont sévères. l’akinésie ou le blocage moteur en période “ off ” sont massifs (stade 5), empêchant la marche ainsi que toute autre activité, alors que les périodes “ on ” restent souvent de bonne qualité (stade 2).
Akinésies circadiennes
Les akinésies circadiennes sont des périodes d’inefficacité thérapeutique de 1 à 3 heures, survenant à horaire fixe, indépendamment de l’horaire des prises de L-dopa, plus volontiers l’après-midi entre 15 heures et 17 heures.
Akinésie nocturne
L’akinésie nocturne s’installe pendant la nuit, 4 à 5 heures après la dernière prise de L-dopa du soir. Le patient est réveillé par des crampes vers 2 heures du matin, il se retourne très difficilement dans son lit.
Akinésie du réveil
L’akinésie du réveil est présente dès le matin, avant la première prise de L-dopa, rendant le lever difficile ou impossible.
Fluctuations non liées aux prises de la L-dopa :
Effet “ on-off ”
L’effet “ on-off ” concerne 15 % à 20 % des cas.
Les fluctuations sont brutales, intenses et très contrastées, passant en 1 minute d’une akinésie majeure à une motricité quasi normale.
Ces effets sont appelés “ on-off ” par les auteurs anglo-saxons du fait de leur brutalité. Il s’agit de fluctuations aléatoires, imprévisibles, très invalidantes d’autant que les périodes “ off ” peuvent s’accompagner de douleurs musculaires, d’anxiété, de sentiments dépressifs, d’oppression thoracique, de dyspnée, de sudation, de pollakiurie.
Akinésies paroxystiques
Les akinésies paroxystiques ou “ freezing ” se manifestent comme un accès d’akinésie brutal, bref et parfois imprévisible, figeant le patient sur place :
– elles peuvent provoquer un piétinement et sont responsables de chutes en avant, chutes “ en statue ”, les pieds restant comme collés au sol.
– décrites avant la L-dopa (G. Lewy, 1922), elles sont souvent déclenchées par une émotion, un obstacle (un tapis, le passage d’une porte) ou un événement inattendu, même familier comme la sonnerie du téléphone.
MOUVEMENTS ANORMAUX :
Les mouvements anormaux représentent une complication particulière à la maladie de Parkinson.
Ils apparaissent en moyenne 3 à 4 ans après le début de la prise de L-dopa et intéressent, après 7 ans de traitement, trois patients sur quatre.
Ils sont provoqués par la L-dopa et par les agonistes dopaminergiques : bromocriptine, lisuride, piribédil, apomorphine.
Aspects cliniques
Sur le plan clinique, les mouvements anormaux ressemblent au mouvement choréique : ils sont brefs, vifs, sans finalité, peu stéréotypés :
– ils parasitent le mouvement volontaire surtout s’ils prédominent à la racine.
– parfois à type de dystonie, ils figent un membre dans une attitude anormale, en extension et en rotation parfois douloureuse.
– leur topographie est fixe pour chaque patient, ils prédominent du côté le plus atteint : à l’extrémité des membres, aux racines, à la face, au cou ou au tronc.
– les anticholinergiques, l’amantadine et le Déprényl ne les font pas apparaître.
Survenue
Leur survenue est liée chronologiquement à la prise de L-dopa :
– les mouvements anormaux de “ milieu de dose ” apparaissent 1 à 3 heures après la prise. lorsque les concentrations plasmatiques sont élevées, ils accompagnent les périodes “ on ”. Ils sont le témoin d’un surdosage dopaminergique mais parfois leur apparition est souvent indissociable de la levée de l’akinésie (période “ on ”) posant de difficiles problèmes thérapeutiques. Le patient étant soit “ on ” avec des mouvements anormaux soit “ off ”.
– les mouvements anormaux de “ début et de fin de dose ” ou biphasiques apparaissent au début et à la fin de la période “ on ”. L’idéal est d’éviter au patient les passages de “ off ” à “ on ” et inversement, il faut alors parfois augmenter les doses de L-dopa afin de prolonger le plus possible la période “ on ”.
Dystonie
Une dystonie peut survenir en. chez d’autres patients, elle survient au petit matin vers 6 heures avant la première prise de L-dopa qui la fait disparaître (“ early morning dystonia ”). Ces dystonies, souvent douloureuses, touchent le côté le plus atteint.
PERTE D’EFFICACITE DU TRAITEMENT :
Il s’agit de la réapparition de la symptomatologie parkinsonienne, en particulier l’akinésie et l’hypertonie, malgré un traitement adéquat qui se révélait efficace les années précédentes :
– ce déclin d’efficacité se distingue des résistances primaires à la L-dopa qui, elles, se constatent d’emblée (et remettent en cause le diagnostic de maladie de Parkinson idiopathique). Il s’installe après plusieurs années de suivi.
– il s’agit de patients généralement âgés ayant une maladie évoluée.
– la période du déclin d’efficacité est annoncée par des chutes.
– des déformations articulaires touchant les extrémités sont souvent associées : “ main d’écrivain ”, “ main de fakir ”, main “ pseudo-rhumatismale ” avec coup de vent cubital, pieds en équin ou le tronc avec cyphose dorsale ou inclinaison latérale.
– l’akinésie et l’hypertonie prédominent nettement au niveau des membres inférieurs, rendant la marche et la station debout impossibles, alors que la motricité des membres supérieurs est relativement préservée.
– des troubles de la déglutition gênent l’alimentation et la prise de traitement et exposent au risque de fausses routes.
– des accès confusionnels ou une détérioration intellectuelle sont souvent présents.
TROUBLES PSYCHIQUES :
Survenue d’accès confusionnels
La survenue d’accès confusionnels ou d’une authentique détérioration intellectuelle est plus liée à l’âge des patients qu’à la durée de l’évolution de la maladie ou du traitement.
Les accès confusionnels ont souvent un facteur déclenchant : fièvre, déshydratation, progression posologique trop rapide, réaction individuelle à un traitement antiparkinsonien.
Presque toujours régressif, cet épisode, s’il se renouvelle, est de mauvais pronostic car il peut annoncer le début d’une détérioration intellectuelle.
Détérioration intellectuelle
La détérioration intellectuelle (10 à 20 % des patients), observée au cours de la maladie de Parkinson idiopathique, évolue lentement, porte surtout sur les troubles mnésiques, l’orientation dans le temps et les difficultés d’attention, l’orientation dans l’espace et le jugement étant assez longtemps conservés.
Il n’existe pas d’apraxie, d’aphasie ou d’agnosie comme dans la maladie d’Alzheimer.
Traitement
Le traitement de la maladie de Parkinson idiopathique doit être adapté à chaque patient, selon les signes cliniques, la gêne ressentie, son âge.
MÉDICAMENTS ANTIPARKINSONIENS :
L-dopa :
L’activité thérapeutique nette, parfois spectaculaire, de la L-dopa s’observe chez 85 % des patients, dès les premières semaines de traitement.
Akinésie et hypertonie
L’akinésie et l’hypertonie sont les premiers symptômes influencés, tandis que l’amélioration du tremblement est plus tardive et parfois incomplète.
L-dopa associée
La L-dopa est associée à un inhibiteur de la décarboxylase périphérique : le bensérazide (Modopar) ou la carbidopa (Sinemet) :
– cet inhibiteur permet d’éviter la dégradation de la L-dopa en dopamine dans le sang circulant.
– il réduit ainsi les effets secondaires périphériques. Comme cet inhibiteur ne passe pas dans le tissu cérébral, il laisse intacte la transformation de la L-dopa en dopamine au niveau de la voie nigrostriée.
Présentations :
– L-dopa et bensérazide (Modopar*) :
– gélules de Modopar* 62,5, 125, 250.
– gélules à libération prolongée : Modopar* 125 LP.
– forme buvable en comprimés : Modopar* 125 Dispersible.
– L-dopa et carbidopa (Sinemet*) :
– comprimés de Sinemet* 100 et 250.
– comprimés à libération prolongée : Sinemet* 200 LP.
Posologie :
– traitement initial : 50 mg trois fois par jour. progressif. faire un palier de 1 à 2 mois et augmenter si nécessaire à 100 mg trois fois par jour.
– la posologie sera ensuite augmentée de 50 à 100 mg par paliers d’au moins 1 mois selon une méthode “ low and slow ”.
– l’adaptation sera progressive et personnalisée. Une fois la dose efficace atteinte, on recherchera par une légère diminution la dose minimale efficace.
Les contre-indications sont rarement formelles :
– ulcères duodénaux ou gastriques en évolution.
– existence de troubles du rythme, d’infarctus myocardique récent et insuffisance cardiaque non stabilisée.
– troubles psychiques : psychose en évolution, détérioration intellectuelle importante, antécédents récents d’accès confusionnels. Le risque d’aggravation sous traitement est alors important.
– la présence de mélanomes impose leur surveillance et leur exérèse à la moindre modification.
– grossesse.
Interactions médicamenteuses
– Les amines sympathomimétiques doivent être évitées. En cas d’anesthésie générale, il est conseillé de cesser le traitement la veille et de le reprendre le lendemain.
– Les IMAO non sélectifs sont déconseillés au risque de provoquer des variations tensionnelles. Par contre la sélégiline (Déprényl*), inhibiteur sélectif de la MAO B, peut être un adjuvant du traitement par la L-dopa.
– Antihypertenseurs à action centrale et réserpine.
– Les neuroleptiques bloquent les récepteurs dopaminergiques et antagonisent ou inversent toutes les actions pharmacologiques de la dopamine.
– La vitamine B6 peut diminuer l’effet thérapeutique de la L-dopa en activant la décarboxylase périphérique.
Les effets secondaires sont peu fréquents :
– nausées, vomissements, gastralgies.
– hypotension orthostatique.
– troubles du rythme.
– confusion, délire.
– mouvements anormaux.
Agonistes dopaminergiques :
Les agonistes dopaminergiques se fixent sur les récepteurs dopaminergiques postsynaptiques et miment toutes les actions pharmacologiques de la dopamine endogène.
Ils évitent ainsi les étapes de synthèse, libération, catabolisme de la dopamine pour agir directement sur les récepteurs postsynaptiques.
Ils sont actifs sur les trois signes parkinsoniens.
Ces produits ont une longue durée d’action qui permet d’atténuer les stimulations dopaminergiques itératives en rapport avec les variations des taux de la L-dopa plasmatique.
Les variations brutales de stimulation des récepteurs sont peut-être un des facteurs favorisant les fluctuations et les mouvements involontaires.
Ils s’adressent :
– aux patients en début de traitement.
– aux patients dont les signes sont insuffisamment contrôlés par la L-dopa.
– aux patients présentant des fluctuations d’efficacité dans la journée.
– aux patients présentant des mouvements anormaux.
Il existe 5 molécules commercialisées : la bromocriptine (Parlodel), le lisuride (Dopergine), le ropinirole (Requip), le piribédil (Trivastal) et, à part, l’apomorphine injectable (Apokinon).
Bromocriptine
Dérivée de l’ergot de seigle, la bromocriptine est connue depuis longtemps pour freiner la sécrétion de prolactine.
Sa durée d’action est d’environ 4 heures.
Présentation : comprimés dosés à 2,5 mg, gélules dosées à 5 et 10 mg.
Posologie :
– de 10 mg à 40 mg. Certains auteurs utilisent de fortes doses en monothérapie (60 mg/j).
– elle sera très progressive : 1/2 comprimé le premier jour au repas du soir, 1 comprimé le deuxième jour, puis augmentation progressive, par palier de 1 comprimé tous les deux jours (en trois prises quotidiennes).
– elle peut être prescrite en monothérapie au début de la maladie et en association à la L-dopa.
Effets secondaires :
– nausées, fréquentes en début de traitement.
– hypotension artérielle moins fréquente, plus gênante, parfois observée dès la première prise, due aux propriétés alpha-bloquantes du produit.
– troubles psychiques à type de confusion mentale, d’hallucinations ou de délire systématisé.
– des précautions sont nécessaires en cas d’ulcère gastro-duodénal et de troubles cardio-vasculaires récents.
– somnolence, une sécheresse de la bouche, constipation, un œdème des membres inférieurs, une pâleur des extrémités chez des patients présentant une sensibilité particulière aux dérivés de l’ergot de seigle (antécédents de syndrome de Raynaud).
Spécifique à la bromocriptine :
– un épanchement pleural a été signalé pour des fortes doses.
– les macrolides (triacétyloléandomycine, érythromycine, josamycine) peuvent augmenter de façon incontrôlable les taux plasmatiques de bromocriptine.
Des nausées ou une hypotension orthostatique peuvent nécessiter la prescription de dompéridone (Motilium*) à la dose de 1 ou 2 comprimés trois fois par jour.
Ropinirole
Le ropinirole (Requip*), agoniste dopaminergique non dérivé de l’ergot de seigle, a été récemment commercialisé.
Sa demi-vie d’élimination est de 6 heures.
Présentation :
– comprimés de Requip* dosés à 0,25 mg, 0,5 mg, 1 mg, 2 mg et 5 mg
– il peut être prescrit en monothérapie au début de la maladie et en association à la L-dopa.
– débuter à 0,25 mg trois fois par jour la première semaine, augmenter progressivement jusqu’à 3 à 9 mg en trois prises sur 2 mois.
Les effets secondaires sont superposables à ceux des agonistes dopaminergiques.
Lisuride
Le lisuride (Dopergine) est un agoniste D2.
Présentation : comprimés de Dopergine dosés à 0, 2 et 0, 5 mg.
Prescrit en association à la L-dopa.
Posologie : débuter à 0,1 mg/j. Augmenter progressivement jusqu’à 0, 8 mg-1 mg en quatre prises sur 2 mois.
Les effets secondaires sont superposables à ceux de la bromocriptine.
Piribédil
Le piribédil (Trivastal) est un agoniste dopaminergique qui présente une action particulière sur le tremblement.
Ce traitement s’adresse aux formes tremblantes : il est utilisé soit seul, comme traitement de première intention, soit associé à la L-dopa.
Il présente une alternative aux anticholinergiques lorsque ceux-ci sont contre-indiqués ou lorsqu’ils doivent être remplacés.
Présentation : comprimés de Trivastal* dosés à 20 mg et 50 mg (à libération contrôlée).
Posologies : doses progressives sont de 100 à 200 mg/j en monothérapie ou en association à la L-dopa.
Les effets secondaires sont pour la plupart identiques à ceux des agonistes :
– nausées, vomissements, hypotension au début du traitement.
– troubles psychiques : état confusionnel, hallucinations visuelles, délire.
– prudence en cas d’affection cardio-vasculaire, d’ulcère gastro-duodénal, de troubles psychiques et d’hypotension artérielle.
Apomorphine
L’apomorphine (Apokinon*) est un agoniste dopaminergique puissant car il a une forte affinité avec les récepteurs dopaminergiques.
Utilisable uniquement par voie sous-cutanée, ce traitement rend service aux patients qui souffrent de périodes “ off ”, peu sensibles aux prises orales de L-dopa ou d’horaire imprévisible.
Le délai d’action est en moyenne de 10 minutes pour une durée de 20 à 45 minutes.
Présentation : Apokinon*, stylo injecteur jetable de 30 mg, aiguilles sous-cutanées de 28 G.
Posologie : injection sous-cutanée de 1 à 9 mg à adapter individuellement.
Pour initier le traitement, un prétraitement de 3 jours par Motilium* est indispensable pour “ antagoniser ” les effets secondaires dopaminergiques : nausées, vomissements, hypotension artérielle.
La dose efficace sera recherchée très progressivement.
Inhibiteurs de la monoamine oxydase de type B :
La sélégiline (Déprényl) est un inhibiteur de la monoamine oxydase de type B (IMAO-B).
Cet inhibiteur augmente la quantité de dopamine disponible au niveau de la synapse dopaminergique.
Son efficacité antiparkinsonienne, modérée, permet de réduire légèrement les doses de L-dopa et peut aplanir les fluctuations de fin de dose débutantes.
Récemment, le Déprényl a retenu l’attention, car il ralentirait peut-être la progression de la maladie de Parkinson. Chez l’animal, il prévient le syndrome parkinsonien induit par le méthyl-phényl-tétrahydropyridine. Par ailleurs, la sélégiline diminue l’oxydation enzymatique de la dopamine par la MAO B, réduisant peut-être ainsi la formation de radicaux libres potentiellement toxiques. Des études récentes pour tenter de vérifier le bien-fondé de cette hypothèse ne l’ont pas confirmé.
Présentation : comprimés dosés à 5 mg.
Posologie : 1 comprimé le matin et à midi, seul en première intention, ou en association à la L-dopa dont il permet parfois de réduire les doses.
Effets secondaires : troubles psychiques (confusion) et hypotension orthostatique.
Interactions médicamenteuses :
– interdit avec la réserpine, les IMAO non sélectifs, la fluoxétine (Prozac*).
– en cas d’anesthésie (moclobémide).
Inhibiteurs de la catéchol-O-méthyl-transférase :
Deux inhibiteurs de la catéchol-O-méthyl-transférase (ICOMT) sont en cours d’autorisation de mise sur le marché : le tolcapone (Tasmar) et l’entacapone.
Ces deux molécules bloquent la transformation de L-dopa en 3-O-méthyl-dopa.
Protégeant la dégradation de la L-dopa, ils prolongent sa demi-vie plasmatique (augmentation de la durée d’effet de chaque prise), sans augmenter les concentrations maximales (pas de surdosage donc pas d’augmentation des mouvements anormaux). Le tolcapone possède un effet périphérique et central.
Ils seront prescrits en association avec la L-dopa, leur meilleure indication sera les fluctuations d’efficacité liées aux prises de L-dopa (akinésies de fin de dose, nocturne ou matinale). Leur effet aboutit schématiquement à celui d’une “ L-dopa retard ”, mais probablement plus facile à manier (absence d’allongement du délai d’action en début de prise).
La posologie est de trois prises par jour.
Interactions médicamenteuses : les IMAO non sélectifs.
Anticholinergiques :
Les anticholinergiques, en entrant en compétition avec l’acétylcholine endogène, bloquent les récepteurs cholinergiques et freinent l’hyperfonctionnement des neurones cholinergiques du striatum.
Cette inhibition s’exerce sur toutes les synapses cholinergiques de l’organisme qu’elles soient centrales ou périphériques (constipation, sécheresse de la bouche). Ils ont été utilisés par Charcot, vers 1870, qui avait remarqué l’effet favorable de la belladone sur le tremblement parkinsonien. Ils sont particulièrement efficaces sur le tremblement.
Les posologies varient selon les composés.
Le plus utilisé en neurologie est le trihexyphénidyle : Artane* 2 et 5 mg, (l’Artane* 15 mg est réservé comme correcteur des syndromes extra-pyramidaux dus aux neuroleptiques).
Effets secondaires :
– sécheresse buccale, constipation, troubles de l’accommodation, dysurie. L’augmentation de la fréquence cardiaque est peu observée.
– plus préoccupant est la survenue de troubles mnésiques ou d’un état confusionnel, le “ délire atropinique ” qui survient surtout chez le sujet âgé même à très faible dose :
– interrompre le traitement très progressivement chez le sujet âgé ou dénutri.
Contre-indications formelles :
– glaucome à angle fermé, l’adénome prostatique, une détérioration intellectuelle ou des accès récents de confusion mentale.
– éviter d’utiliser après 65 ans, contre-indiqué en présence de troubles mnésiques.
Amantadine :
Le chlorhydrate d’amantadine (Mantadix) est utilisé depuis les années 60 comme antiviral (virus grippaux du groupe A). En 1969, Schwalbe le découvre de façon empirique.
L’action antiparkinsonienne du produit et son mécanisme d’action sont encore inconnus (action antiglutamatergique ?).
Avant traitement, rechercher une insuffisance rénale qui évitera la survenue des effets secondaires : œdème des membres inférieurs avec livédo reticularis, hallucinations visuelles, confusion.
Présentation : gélules dosées à 100 mg.
Posologie : de 200 à 400 mg/j.
L’action de l’amantadine améliore surtout l’akinésie.
Chez quelques patients son efficacité est spectaculaire, permettant une motricité satisfaisante sur une longue période en monothérapie.
Sa principale indication est en début de maladie en monothérapie. Elle peut aussi être prescrite en association à la L-dopa car elle a l’avantage de ne pas provoquer ou accentuer les mouvements anormaux.
Régime pauvre en protéines :
Un régime pauvre en protéines a été proposé dans le but de limiter l’apport alimentaire en acides aminés aromatiques.
Ces acides aminés peuvent entrer en compétition avec la L-dopa au niveau du passage de la barrière digestive, mais aussi intracérébrale ou intraneuronale.
Ce régime est proposé principalement au repas de midi, car les défauts d’absorption de L-dopa se produisent le plus souvent dans l’après-midi.
Il est relativement astreignant et ne doit être prolongé que lorsque son efficacité a été démontrée, car certains patients sont insensibles à de tels régimes et ce dernier peut entraîner un amaigrissement préjudiciable.
CONDUITE DU TRAITEMENT :
Les indications et les posologies doivent correspondre aux données du Vidal.
Le but est de maintenir, à long terme, le bénéfice thérapeutique obtenu au début du traitement en sachant que la maladie continue à évoluer sous traitement.
Il faut proposer au patient “ le meilleur confort, le plus longtemps possible ” (A. Guillard).
En dehors des anticholinergiques et de l’amantadine (mécanisme d’action inconnu), tous les antiparkinsoniens visent à restaurer le fonctionnement de la voie dopaminergique nigrostriée.
Traitement initial et pendant la “ lune de miel ” :
Le début du traitement se décidera sur la gêne fonctionnelle.
Un patient peu gêné peut ne pas être traité et sera revu 2 mois plus tard. Cela permet d’avoir une idée sur l’évolution spontanée de l’affection.
Traitement en première intention
– En première intention, si la gêne est peu importante, on peut utiliser un antiparkinsonien “ mineur ” : amantadine ou anticholinergique.
– Sinon débuter le traitement soit par la L-dopa, soit par un agoniste dopaminergique, mais pas par les deux à la fois (pour apprécier l’efficacité et la tolérance respective des deux produits).
– Le patient sera ensuite maintenu, soit sous L-dopa, soit sous agoniste dopaminergique tout au long de la “ lune de miel ”.
Alternative
L’alternative est de réaliser une association précoce L-dopa-agoniste : une fois le traitement stabilisé, lors du premier fléchissement d’effet, plutôt que d’augmenter la dose, ajouter soit un agoniste à la L-dopa soit l’inverse :
– de cette façon, le patient se trouve rapidement (1 an) traité par une association précoce L-dopa-agoniste :
– cela permet, pour des doses moindres de chaque produit prescrit seul, d’avoir une meilleure efficacité (effet synergique).
– cette association précoce permettrait, en outre, de retarder les fluctuations d’efficacité et les mouvements anormaux mais cela n’est pas formellement prouvé.
Le traitement à ce stade est relativement simple, et les adaptations posologiques se limitent à la recherche des doses minimales efficaces ainsi qu’à une bonne tolérance.
Traitement des complications :
Fluctuations
– Lorsque les fluctuations sont peu intenses, la première mesure peut consister à ajouter la sélégiline, qui peut suffire à les faire disparaître. Lorsque les fluctuations atteignent une certaine intensité, la mesure la plus fréquemment utilisée est de répartir les prises de L-dopa dans la journée en augmentant modérément la dose journalière, en passant de quatre à cinq prises par jour.
– Ensuite, on peut faire appel aux agonistes dopaminergiques :
– la bromocriptine, agoniste dopaminergique de référence, prescrite à doses très lentement progressives de 10 à 40 mg/j, atténue très sensiblement les fluctuations.
– l’emploi d’un autre agoniste dopaminergique, le lisuride ou le ropinirole, obéit au même schéma et le piribédil donne des résultats intéressants surtout lorsque subsiste un tremblement de repos.
– Une autre approche consiste à utiliser les formes de L-dopa à libération prolongée (Modopar LP 125 et Sinemet LP 200). Prescrites chez les patients qui ont des fluctuations régulières, elles allongent très nettement les périodes.
Akinésie de l’après-midi
Pour l’akinésie de l’après-midi, utiliser le Modopar 125 Dispersible pour la prise de l’après-midi, cette forme buvable de L-dopa est alors mieux absorbée.
Akinésie nocturne
En ce qui concerne l’akinésie nocturne, une prise de L-dopa à libération prolongée au coucher ou dans le milieu de la nuit est généralement très efficace. Cette prise est souvent efficace sur l’akinésie du réveil.
Faire remplir un carnet de surveillance des fluctuations. La coopération du patient ou de l’entourage est nécessaire.
Traitement à la phase de déclin :
Au stade de perte d’efficacité et de troubles psychiques que représente la phase de déclin, l’acharnement thérapeutique n’est pas de règle.
Autant il est justifié dans les fluctuations d’efficacité, une modification apparemment minime apportant parfois une amélioration sensible, autant lorsqu’il existe des troubles psychiques, la tolérance des traitements ne permet pas d’atteindre la posologie efficace.
Il faut, devant une telle situation, s’abstenir de prescrire des anticholinergiques, des antidépresseurs tricycliques et de l’amantadine.
Les agonistes dopaminergiques seront maniés avec la plus grande prudence.
Il reste alors la L-dopa qui demeure la mieux tolérée, à posologie modérée (250 mg/j).
Cas particuliers
Dans les cas particuliers, les troubles neuro-végétatifs, l’anxiété, la détérioration intellectuelle et les troubles articulaires seront traités.
RÉÉDUCATION FONCTIONNELLE :
Les malades atteints de maladie de Parkinson peuvent bénéficier d’une rééducation.
Celle-ci améliore l’autonomie, la gêne motrice ainsi que les complications orthopédiques et respiratoires de l’affection. Le malade conserve plus longtemps son autonomie. Les techniques sont différentes en fonction du degré de l’atteinte.
La kinésithérapie sera prescrite dès que le malade présente des signes francs de rigidité ou d’akinésie, pour lutter tôt contre l’apparition des déformations articulaires.
Dans les formes plus évoluées, la rééducation est centrée sur la marche, la prévention des chutes.
Au stade très évolué, elle maintiendra les activités de la vie quotidienne et les transferts.

")





")


")
{kind=link}