



















Introduction :
La mucine est une substance amorphe d’aspect gélatineux qui est une composante normale des organes humains.
Deux types de mucine sont connus, l’un épithélial et l’autre dermique, qui ont une structure différente et qui se colorent différemment.
La mucine dermique est trouvée dans la substance fondamentale du derme et est produite normalement en petites quantités par les fibroblastes.
 La mise en évidence de la mucine dermique nécessite des colorations spéciales des coupes histologiques.
La mise en évidence de la mucine dermique nécessite des colorations spéciales des coupes histologiques.
Le bleu Alcian à pH 2,5 la colore en bleu et le fer colloïdal en bleu-vert. De plus, elle est sensible à la hyaluronidase.
Cette mucine dermique est composée d’acide hyaluronique et de dermatane sulfate, liés à des petites quantités de chondroïtine-6-sulfate et de chondroïtine-4-sulfate.
Elle est très hydrophile et capable d’absorber 1 000 fois son poids en eau.
Elle joue par conséquent un rôle important dans l’équilibre sel/eau, remplissant les espaces intercellulaires et aidant la migration des éléments chimiques et des cellules dermiques.
La mucine épithéliale, au contraire, est présente dans le tube gastrodigestif et les poumons.
Dans la peau, elle se retrouve dans les cellules sombres des glandes eccrines et dans les cellules apocrines.
Elle est constituée par des glycosaminoglycanes neutres et acides, est colorée par le bleu Alcian à pH 2,5, comme la mucine dermique, mais est PAS (acide périodique Schiff)-positive, résistante à la hyaluronidase et n’est pas métachromatique avec le bleu de toluidine.
Les mucinoses cutanées sont un groupe hétérogène de maladies au cours desquelles la mucine dermique s’accumule dans le derme et dans les follicules pileux.
Les mucopolysaccharidoses, affections où la mucine déposée est constituée par la chondroïtine sulphate, ne sont pas traitées dans ce chapitre.
Classification :
Dans l’attente que l’étiopathogénie des mucinoses cutanées soit éclaircie, celles-ci peuvent être divisées en deux groupes provisoires : les mucinoses cutanées spécifiques (primaires) se manifestant par des lésions cliniques spécifiques ou le dépôt de mucine est le signe histologique distinctif, et les maladies cutanées où le dépôt de mucine est simplement un signe histologique accessoire (mucinoses secondaires).
Selon la localisation microscopique de la mucine, le premier groupe, celui des mucinoses spécifiques, peut être divisé en mucinoses dermiques et folliculaires.
En revanche, le second groupe est divisé en mucinoses épidermiques, dermiques et folliculaires.
Ce chapitre envisage les aspects cliniques, histopathologiques et le traitement des seules mucinoses cutanées spécifiques (primaires) dont la classification est également remise à jour.
Les mucinoses cutanées spécifiques se groupent en mucinoses inflammatoires/dégénératives et en mucinoses néoplasiques/hamartomateuses.
Le premier groupe comprend les mucinoses dermiques et les mucinoses folliculaires.
Mucinoses spécifiques (primaires) inflammatoires/dégénératives :
A – MUCINOSES DERMIQUES :
1- Lichen myxoedémateux ou mucinose papuleuse :
C’est une maladie chronique qui se caractérise par une éruption papuleuse due au dépôt dermique de mucine et à un variable degré de prolifération fibroblastique sans anomalies thyroïdiennes associées.
Il y a deux types principaux de lichen myxoedémateux (LiM) selon leur extension et la présence ou pas d’une atteinte systémique : la forme généralisée et sclérodermoïde appelée aussi scléromyxoedème, et la forme localisée.
La progression d’une forme à l’autre a été rapportée, mais jamais prouvée.
Le scléromyxoedème est une affection rare qui touche les sujets d’âge moyen.
Hommes et femmes sont également atteints.
Les lésions typiques consistent en des papules lichénoïdes d’allure cireuse de 3 à 6 mm de diamètre, sur un fond infiltré, érythémateux et sclérodermoïde, qui peut atteindre le tégument presque dans sa totalité, avec un perte de mobilité, en particulier de la bouche et des doigts.
L’épaississement et les rides longitudinales de la glabelle, tout à fait typiques, peuvent donner au visage un aspect léonin.
La plupart des observations de scléromyxoedème (83 %) sont associées à une gammopathie monoclonale, généralement du type 7S-immunoglobuline (Ig)G lambda.
L’évolution vers un authentique myélome n’est pas fréquente (10 % des cas).
Les autres manifestations systémiques associées au scléromyxoedème incluent des atteintes myositiques, neurologiques, articulaires, pulmonaires, cardiovasculaires et rénales. Dix cas de coma sans explication organique ont été signalés.
L’évolution du scléromyxoedème est imprévisible.
Le pronostic peut être mauvais et le décès secondaire à une bronchopneumonie, une thrombose vasculaire, plus rarement à l’atteinte spécifique d’un organe ou à la survenue d’un coma.
Le LiM localisé se présente avec des papules mucineuses sans aucune infiltration sclérodermoïde et sans gammopathie monoclonale ni atteinte systémique.
Les papules peuvent rester isolées ou devenir coalescentes pour former des lésions linéaires, des nodules et/ou des plaques sur les extrémités supérieures et inférieures et le tronc.
Il n’y a pas d’autre anomalie biologique dans le LiM localisé, quoique tout récemment l’association à une infection par le virus de l’immunodéficience humaine (VIH) ait été décrite.
De tout façon, le LiM localisé est une forme bénigne purement cutanée.
L’examen histologique du scléromyxoedème montre des dépôts de mucine dans le derme réticulaire associés à des faisceaux de collagène épaissis et à une prolifération fibroblastique.
Les fibres élastiques sont raréfiées et il peut exister un infiltrat périvasculaire lymphoplasmocytaire.
Les dépôts de mucine peuvent toucher d’autres organes comme les coronaires, le rein, le pancréas, les glandes surrénales et les fibres nerveuses.
Dans le LiM localisé, on observe un dépôt de mucine bien circonscrit au derme superficiel et moyen, avec une prolifération variable des fibroblastes sans aucune fibrose.
L’étiologie est inconnue ; un lien avec la gammapathie monoclonale n’a pas été prouvé. Le sérum des patients, même après élution de l’Ig monoclonale, peut stimuler la prolifération fibroblastique.
La stratégie thérapeutique doit être différente pour les deux formes.
Le traitement du scléromyxoedème est décevant et il n’y a pas une thérapie d’élection.
Le melphalan est la thérapie de choix aux États- Unis soit en monothérapie, soit associé aux stéroïdes ou à la plasmaphérèse.
Les effets secondaires, en particulier les infections opportunistes graves et les néoplasies hématologiques, sont un facteur limitant important.
Des autres agents chimiothérapeutiques (cyclophosphamide, méthotrexate, chlorambucil) ou la seule corticothérapie systémique n’ont pas donné de meilleurs résultats. Radiothérapie, électronthérapie, rétinoïdes, plasmaphérèse,
photophérèse extracorporelle, PUVAthérapie ont donné des résultats variables. L’efficacité des stéroïdes et/ou du diméthylsulfoxyde locaux ou de la dermabrasion pour traiter l’inesthétisme a été signalée.
Une régression spontanée, même après 15 ans, a été rapportée. Les traitements agressifs doivent être limités aux patients défigurés, invalides ou ayant des complications systémiques.
Dans le LiM localisé, la thérapie n’est pas nécessaire et une stratégie de surveillance est conseillée.
La corticothérapie locale peut être parfois efficace. Une régression spontanée est possible, même dans la forme associée au VIH.
2- Mucinose érythémateuse réticulée ou mucinose cutanée en plaque (syndrome REM, « midline mucinosis ») :
Elle touche avec prédilection la femme d’âge moyen, quoique les enfants d’âge prépubertaire puissent être affectés.
Elle est caractérisée par des papules et macules érythémateuses, parfois prurigineuses, confluentes en plaque ou d’allure réticulaire ou annulaire dans la zone médiodorsale ou médiothoracique.
L’abdomen peut être parfois atteint. L’exposition solaire exacerbe l’éruption et le prurit.
Des lésions de même aspect ont été reproduites par l’exploration photobiologique.
En général, la mucinose érythémateuse réticulée n’est pas associée à des manifestations systémiques et aucun examen biologique n’est utile.
Cependant, elle a été parfois associée à des cas d’hyper- ou hypothyroïdie, de thyroïdite d’Hashimoto, de lupus chronique, de carcinomes colique et pulmonaire, de purpura thrombopénique, d’anémie réfractaire aux excès des blastes.
Des cas d’aggravation par prise de contraceptif oral, par les règles ou la grossesse ont été également rapportés.
L’examen histologique montre, sous un épiderme qui est toujours normal, des dépôts de mucine, parfois discrets, le long des faisceaux de collagène du derme superficiel.
Il existe un infiltrat périvasculaire, occasionnellement périfolliculaire, formé principalement par des lymphocytes CD4 et des dilatations des capillaires du derme superficiel.
L’immunofluorescence directe est habituellement négative, mais des dépôts granuleux d’IgM, IgA et C3 ont été retrouvés sur la membrane basale.
L’étiologie est inconnue.
L’exposition solaire et des agents viraux ont été incriminés comme facteurs responsables ou déclenchants ; des particules d’allure virale ont été identifiées dans les cellules endothéliales dermiques.
Une relation entre la mucinose érythémateuse réticulée et le lupus érythémateux peut être envisagée.
La mucinose érythématheuse réticulée est considérée comme une pathologie bénigne qui peut persister plusieurs années, parfois audelà de 15 ans.
Les antipaludéens de synthèse restent le traitement de choix généralement efficace en 2 à 4 semaines.
3- Scléroedème :
C’est une induration symétrique de la moitié supérieure du corps précédée par une infection ou associée à un diabète.
Il y a trois types de scléroedème.
– Le premier type est une forme bénigne, précédée par de la fièvre, une fatigue et une infection des voies respiratoires généralement streptococcique.
Les femmes d’âge moyen sont les plus touchées, mais les enfants peuvent l’être aussi.
L’induration de la peau est de survenue brutale, surtout aux régions cervicofaciales et gagnant ensuite la racine des membres supérieures.
Le visage apparaît sans expression, l’ouverture de la bouche et la déglutition sont difficiles du fait d’une atteinte linguale et pharyngienne.
Ce type de scléroedème guérit habituellement en quelques mois.
– Quoique se présentant cliniquement de façon similaire, le deuxième type n’est pas précédé d’une infection aiguë, présente un début plus insidieux et peut persister plusieurs années.
– Le troisième type est associé à un diabète mal contrôlé (scleroedema diabeticorum) et survient surtout chez les hommes obèses, d’âge moyen.
Le début est insidieux et l’affection se prolonge indéfiniment.
Les lésions cutanées ne sont pas modifiées par le traitement du diabète.
Les manifestations systémiques peuvent survenir dans toutes les formes, avec des épanchements pleuraux et péricardiques, des dysarthries et des dysphagies, des anomalies oculaires, des myosites, des atteintes cardiaques, des parotidites.
Les associations avec une dysglobulinémie monoclonale, une polyarthrite rhumatoïde, un syndrome de Gougerot-Sjögren, une hyperparathyroïdie primaire, un insulinome malin ont été rapportés.
L’examen histologique montre un épaississement dermique avec des faisceaux de collagène oedémateux, séparés par des dépôts de mucine qui sont parfois discrets ou difficiles à mettre en évidence.
Le tissu graisseux est réduit et probablement remplacé par du collagène.
La mucine peut aussi être retrouvée dans les muscles striés ou le myocarde.
L’étiologie est inconnue. Une « hypersensibilité streptoccocique », un traumatisme des vaisseaux lymphatiques, l’Ig monoclonale et le diabète pourraient jouer un rôle.
Le scléroedème s’accompagne en fait d’une morbidité peu importante à part la limitation des mouvements.
Le type I peut guérir en 6 mois à 2 ans, tandis que les autres types persistent plusieurs années.
Dans le type III, la régression est difficile et des décès ont même été rapportés.
Il n’existe pas de traitement spécifique. Les corticoïdes systémiques et intralésionnels, le méthotrexate, l’exposition ultraviolette, les antibiotiques et la pénicillamine n’influencent pas l’évolution de la maladie.
Les bolus de cyclophosphamide et la prednisolone par voie orale seraient efficaces.
La ciclosporine a été aussi utilisée avec succès.
Dans le scléroedème associé au myélome, la chimiothérapie pour la néoplasie hématologique a amélioré la dermatose.
4- Mucinoses des thyroïdiens :
Ce groupe comporte le myxoedème localisé, le myxoedème généralisé et la mucinose papuleuse associée aux maladies thyroïdiennes.
* Myxoedème localisé (prétibial) :
Le myxoedème prétibial est une induration de la région prétibiale associée à une hyperthyroïdie ou survenant après thyroïdectomie.
Il s’agit d’un des cinq signes de la maladie de Basedow (0,4-5 % d’incidence) avec le goitre, l’exophtalmie, l’acropathie thyroïdienne et l’élévation du long acting thyroid stimulator (LATS).
Plus rarement, il peut survenir dans le cadre d’une thyroïdite de Hashimoto et sans thyrotoxicose, lors de l’hypothyroïdie, se développant après traitement de la maladie de Basedow et aussi chez des patients euthyroïdiens.
Le myxoedème localisé apparaît sous la forme de plaques ou de nodules, érythémateux bruns, pourpres ou jaunes, cireux, indurés ou en « peau d’orange ».
En général, les lésions sont localisées sur les faces antérolatérales des jambes ou des pieds.
Le myxoedème localisé peut aussi apparaître sous l’aspect d’un oedème diffus des jambes qui évolue vers un état éléphantiasique.
Plus rarement, le myxoedème localisé touche la face, les épaules, les extrémités supérieures et l’abdomen ou les cicatrices.
Les lésions sont parfois douloureuses et prurigineuses. Une hypertrichose et une hyperhidrose localisées dans la peau myxoedémateuse ont été rapportées.
Histologiquement, il y a de larges dépôts de mucine dans le derme réticulaire et l’hypoderme séparant les faisceaux de collagène et entourant les glandes eccrines.
Un infiltrat lymphocytaire discret périvasculaire et périannexiel s’associe à des mastocytes et à de larges fibroblastes étoilés.
Les fibres élastiques sont réduites.
La morbidité est faible, mais l’engainement des nerfs péroniers par la mucine peut entraîner un pied tombant ou une gêne à la dorsiflexion.
L’étiologie est inconnue. Un facteur sérique (sans liens avec le LATS) stimulerait la production par les fibroblastes d’une grande quantité de mucine.
Les traumatismes, un facteur insuline-like, une obstruction lymphatique pourraient jouer aussi un rôle.
Le traitement n’est pas toujours satisfaisant. Les corticoïdes sous occlusion ou intralésionnels peuvent être efficaces.
La chirurgie a été rapportée comme étant efficace mais des récidives ont suivi des greffes cutanées.
L’élastocompression ou la compression pneumatique de la jambe ont été utilisées avec succès.
Le traitement de la maladie de Basedow ou des autres causes n’entraîne aucun bénéfice.
Il faut aussi néanmoins signaler des régressions spontanées.
* Myxoedème généralisé :
C’est une manifestation d’hypothyroïdie grave due à l’accumulation généralisée de la mucine dans le derme et dans d’autres tissus.
L’hypothyroïdie peut être congénitale (crétinisme), juvénile ou acquise.
La forme congénitale (1 cas sur 5 000 naissances) doit être évoquée dès les premiers mois lorsqu’il y a une somnolence, une constipation, des difficultés d’alimentation, une hypotonie musculaire, la persistance d’un ictère et des troubles respiratoires.
Le myxoedème généralisé (MG) est responsable d’un oedème généralisé ne prenant pas le godet et donnant un aspect typique d’oedème périorbitaire, des lèvres, de la langue, des régions acrales et génitales.
La peau est sèche, froide et pâle, les cheveux sont secs et cassants avec des zones d’alopécie, les ongles sont fragiles.
La présence d’un bourrelet claviculaire peut aussi évoquer le diagnostic. La survenue d’hypothyroïdie chez un enfant est appelée « hypothyroïdie juvénile ».
Les signes sont caractérisés par une petite taille, un développement physique et mental anormal, un retard de la maturité sexuelle et un faible niveau scolaire.
Certains enfants développent une hypertrichose des épaules et de la partie haute du dos.
L’hypothyroïdie de l’adulte est la forme la plus commune.
Habituellement, celle ci est observée chez les femmes entre 40 et 60 ans, déclenchée par une thyroïdite d’Hashimoto ou par le traitement d’une maladie de Basedow ; plus rarement, elle peut être la conséquence d’une défaillance hypophysaire (hypothyroïdie secondaire) ou hypothalamique (tertiaire).
La symptomatologie initiale est souvent non spécifique avec une asthénie, une prise de poids, une constipation, une perte d’appétit, une frilosité, des crampes dans les jambes.
Le visage est triste, apathique ; les paupières, les lèvres, la langue et les mains sont oedémateuses, le nez est large et la voix rauque.
La peau est pâle ou jaunâtre, froide, cireuse et sèche.
Les cheveux sont secs et avec une alopécie non cicatricielle, et les ongles sont fragiles.
Les autres signes peuvent comprendre une hypohidrose, un purpura des membres inférieurs, une acrocyanose, un retard à la cicatrisation des plaies, des xanthomes, une cardiomégalie, un mégacôlon, un syndrome du canal carpien ou une paralysie faciale.
L’histologie montre des dépôts dermiques, parfois discrets, de mucine surtout périfolliculaires et périvasculaires.
Les fibres élastiques sont réduites.
Des dépôts cérébraux seraient responsables de manifestations psychiatriques.
Le diagnostic est porté sur l’association des signes cliniques et la diminution de la T3 et T4.
La thyroid stimulating hormone (TSH) est augmentée dans l’hypothyroïdie primaire et basse dans la forme secondaire où le myxoedème généralisé peut être absent.
Tous les signes du myxoedème sont réversibles avec la restauration de la fonction thyroïdienne.
La thérapie substitutive, qui comporte la lévothyroxine, doit être graduelle.
En l’absence de traitement peuvent survenir un coma myxoedémateux et la mort.
Le diagnostic et le traitement précoces sont cruciaux pour le développement mental.
Les dosages de T3 et T4 doivent être effectués entre le troisième et le sixième jour de vie et un traitement substitutif doit être institué avant le quatrième mois.
* Mucinose papuleuse associée à une maladie thyroïdienne :
Une mucinose papuleuse a été décrite associée à une thyroïdite d’Hashimoto et à une hypothyroïdie.
5- Mucinose papulonodulaire au cours du lupus érythémateux :
Également appelée mucinose cutanée lupique, la mucinose papulonodulaire (MPN) survient dans 1,5 % des cas de lupus érythémateux (LE) sous forme de papules ou nodules, couleur peau normale ou érythémateuse, de 0,5 à 2 cm de diamètre donnant à la peau un aspect en « mottes » bien visible « à jour frisant ».
Une dépression centrale et une hyperpigmentation peuvent être caractéristiques.
Des lésions en plaques, dues à un dépôt massif de mucine, ont été aussi observées.
Le tronc, le cou, les membres supérieurs sont électivement atteints.
La MPN peut précéder, débuter avec ou accompagner le LE.
Dans 75 % des cas, il s’agit d’un LE systémique avec atteinte rénale (50 %) et articulaire (40 %).
L’histologie montre un dépôt diffus de mucine dans le derme superficiel et moyen avec parfois un discret infiltrat lymphocytaire périvasculaire, mais sans aspect spécifique histologique de lupus.
Le traitement correspond à celui du LE.
Des injections intralésionnelles de hyaluronidase ont été essayées pour les nodules de grande taille.
6- Mucinose cutanée juvénile spontanément régressive :
Elle survient chez l’enfant avec l’apparition brutale de papules asymptomatiques se rassemblant en plaques linéaires, infiltrées, érythémateuses et donnant une apparence ondulée à la peau.
Les lésions atteignent la face, le cou, le cuir chevelu, le tronc et les cuisses.
Des nodules profonds sont aussi présents sur les articulations, associés à une arthrite des genoux, coudes et doigts.
L’histologie révèle des dépôts de mucine dans le derme réticulaire superficiel avec une augmentation des fibroblastes et des mastocytes.
La mucine est aussi retrouvée dans les nodules périarticulaires.
La mucinose cutanée juvénile spontanément régressive (MCJSR) guérit spontanément.
7- Mucinoses cutanées toxiques :
Ce groupe inclut les mucinoses papuleuses dans le cadre du syndrome des huiles toxiques et du syndrome myalgie-éosinophilie.
Bien que sans relation épidémiologique, le syndrome des huiles toxiques, rapporté en Espagne après ingestion d’huile de colza dénaturée, et le syndrome myalgie-éosinophilie associé à l’utilisation de produits contenant du L-tryptophane, comprennent plusieurs symptômes communs, tels des infiltrats pulmonaires, une asthénie, une éosinophilie périphérique et des manifestations cutanées.
Parmi ces dernières, il peut y avoir une mucinose papuleuse spontanément régressive, des lésions sclérodermiformes ou une fasciite éosinophile.
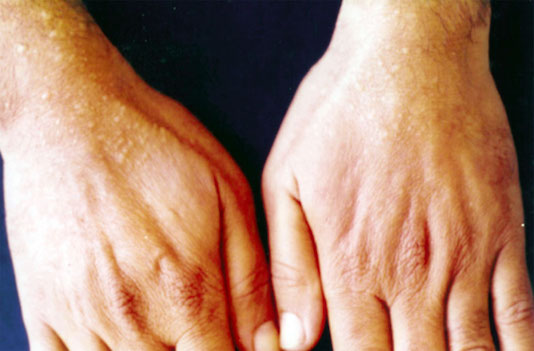
8- Mucinose papuleuse acrale persistante :
La mucinose papuleuse acrale persistante (MPAP) touche principalement les femmes et est caractérisée par des papules multiples, ivoirines ou de couleur chair de 2 à 5mm de diamètre et à disposition symétrique.
Elles sont localisées exclusivement sur les dos des mains et les faces d’extension des poignets.
Les lésions persistent et progressent lentement sans régression. Histologiquement, on retrouve un dépôt de mucine en larges foyers bien circonscrits du derme médiosuperficiel.
Le dépôt est parsemé des fibres de collagène et respecte une zone sous-épidermique.
Le nombre des fibroblastes est normal.
La possibilité que les mucinoses acrales papuleuses soient une présentation particulière du LiM localisé a été en fait l’objet de discussion.
La MPAP est de bon pronostic.
Les applications topiques de stéroïdes ou de hyaluronidase ont été inefficaces.
9- Mucinose cutanée infantile :
Elle consiste en des papules fermes, de 1 à 2mm de diamètre, opalescentes, groupées aux coudes et associées à quelques papules éparpillées, parfois linéaires, sur les dos des mains et les bras.
La maladie est précoce (4 mois de vie) ou même congénitale.
Quelques cas sont considérés comme un nævus mucineux.
La mucine est accumulée d’une façon si superficielle dans le derme qu’elle semble enserrée par l’épiderme.
Il n’y a pas de traitement.
L’éruption ne guérit pas spontanément.
10- Mucinose cutanée focale :
La lésion est une papule ou un nodule à la couleur chair ou blanche, asymptomatique, habituellement unique, de moins de 1 cm de diamètre.
La face, le tronc et les membres sont touchés chez l’adulte à l’exception des articulations des mains et pieds.
Le diagnostic est histologique.
Des dépôts circonscrits mais mal délimités de mucine dermique qui épargnent le tissu sous-cutané sont mis en évidence, ainsi que des fibroblastes fusiformes ou étoilés.
Ces cellules sont marquées par les anticorps antivimentine et sont associées à une population moins représentée de dendrocytes dermiques qui sont en partie marqués par l’antifacteur XIIIa et, en partie, par l’anti-CD34.
Les fibres élastiques et réticulaires sont absentes.
Les capillaires sont normaux. La mucinose focale est considérée comme une lésion réactionnelle qui doit être différentiée de l’angiomyxome.
Le traitement de choix est l’excision chirurgicale.
Il n’y a pas de récidive.
11- Neuropathie cutanée mucineuse :
Un seul cas a été rapporté chez un jeune homme qui a développé des lésions cutanées livédoïdes et réticulées sur les extrémités inférieures avec une hyperesthésie.
L’histologie montrait une hypertrophie des fibres nerveuses entourées de mucine.
12- Kyste mucoïde digital (kyste synovial, kyste myxoïde) :
Le kyste mucoïde digital (KMD) est une lésion fréquente qui apparaît sous la forme d’un nodule kystique, presque translucide, dépassant rarement 2 cm.
Il est classiquement localisé sur la troisième phalange des doigts.
Il y a deux sources principales de mucine : les cellules synoviales et les fibroblastes dermiques.
Les KMD dérivés des cellules synoviales sont localisés au-dessus des articulations et sont dus à une herniation de la cavité articulaire ; en revanche, les KMD dérivés des fibroblastes se retrouvent entre les articulations des phalanges et ne sont pas de vrais kystes, mais résultent d’une dégénérescence mucineuse du tissu conjonctif.
Dans ce cas, l’aspect histologique typique est celui d’un large espace kystique non encapsulé rempli de mucine avec des fibroblastes étoilés.
Le dépôt de mucine peut être délimité par une paroi composée de tissu conjonctif. Parfois, une élimination transépidermique est observée.
Plusieurs traitements chirurgicaux ont été proposés, mais les récidives sont fréquentes.
13- Formes diverses :
Les autres mucinoses primitives dont la nosologie n’est pas claire, comprennent le myxoedème tubéreux atypique de Jadassohn-Doesseker qui correspondrait a une forme de lichen myxoedémateux nodulaire, des mucinoses cutanées à régression spontanée de l’adulte, une mucinose périfolliculaire chez un sidéen.
B – MUCINOSES FOLLICULAIRES :
1- Mucinose folliculaire de Pinkus (alopécie mucineuse) :
Il s’agit d’une maladie inflammatoire caractérisée par des papules folliculaires et des plaques indurées dans lesquelles la mucine s’accumule dans les follicules pilosébacés et aboutit à une alopécie.
Cette forme primitive n’est pas associée à des lymphomes ou autres affections.
Il existe une forme aiguë, qui touche le cuir chevelu et la face chez les enfants, et une forme chronique, plus diffuse au tronc et aux extrémités, qui prédomine chez les adultes d’âge jeune ou moyen.
En plus de la mucine retrouvée dans les follicules pilosébacés, il existe un infiltrat périvasculaire et périfolliculaire de lymphocytes, histiocytes et éosinophiles.
La distinction histologique entre la mucinose folliculaire de Pinkus et la mucinose folliculaire associée à un lymphome est difficile.
Les signes qui plaident en faveur d’une mucinose folliculaire de Pinkus sont l’absence d’infiltrat lymphocytaire épidermotrope et d’atypie lymphocytaire, un dépôt considérable de mucine dans le follicule pilosébacé, un infiltrat inflammatoire périvasculaire et périfolliculaire plutôt que de type diffus ou nodulaire, beaucoup d’éosinophiles, peu de plasmocytes et surtout l’absence d’un réarrangement clonal T.
L’étiologie de la mucinose folliculaire de Pinkus est inconnue et il n’y a pas de traitement de choix.
Une stratégie de surveillance peut être envisagée car la guérison spontanée sans récidive est possible en 2 à 24 mois.
Les corticoïdes topiques ou systémiques et l’interféron alpha ont donné un certain bénéfice.
2- Mucinose folliculaire « urticaria-like » :
Cette mucinose folliculaire, plus fréquente chez l’homme d’âge moyen, se caractérise par des papules ou des plaques prurigineuses et urticariennes de la face et du cou sur un fond d’érythème « séborrhéique ».
Les lésions disparaissent en laissant une macule rouge qui persiste quelques semaines.
L’éruption évolue par poussées irrégulières et peut persister de 2 mois à 15 ans.
Il n’y a pas d’atteinte systémique. Le diagnostic différentiel se pose surtout avec la dermatite séborrhéique, mais l’histologie, qui montre des espaces kystiques remplis de mucine dans les follicules pilosébacés, est distinctive.
Le pronostic est bon.
Le traitement est habituellement décevant, mais les antipaludéens de synthèse peuvent aider.
Mucinoses cutanées spécifiques néoplasiques/hamartomateuses :
A – NÆVUS MUCINEUX :
Le nævus mucineux est un hamartome bénin qui peut être à la fois congénital ou acquis.
Il s’agit d’une plaque papuleuse, habituellement avec une distribution linéaire, unilatérale.
Histologiquement, le dépôt de mucine est diffus dans le derme superficiel. Les fibres collagènes et élastiques ont disparu dans la zone de dépôt de mucine.
L’épiderme est normal ou peut comporter une hyperkératose et une acanthose avec une élongation des crêtes interpapillaires qui rappellent un nævus épidermique.
Dans ce dernier cas, le nævus mucineux peut être un hamartome combiné qui associe des aspects d’un nævus épidermique avec ceux d’un nævus conjonctif du type à protéoglycans.
Sous le terme de « fibrokératomes mucineux acraux » ont été récemment rapportées des lésions kératosiques et mucineuses héréditaires des mains.
B – (ANGIO)MYXOME :
Le myxome cutané est une tumeur bénigne acquise. Les termes « angiomyxome » et « myxome » sont des synonymes et indiquent la même entité.
Le myxome peut être solitaire sans aucune anomalie systémique ou bien multiple. Dans ce dernier cas, il peut représenter une manifestation du complexe de Carney (myxomes cutanés, myxome cardiaque, lentigos, hyperactivité endocrinienne).
Cependant, il y a aussi des myxomes multiples sans les autres éléments du syndrome. Histologiquement, il s’agit d’une matrice mucineuse bien démarquée qui intéresse le derme et le tissu sous-cutané avec des fibroblastes polymorphes, des mastocytes et des fibres collagènes et réticulaires.
On observe aussi des cellules multinucléées bizarres avec des figures mitotiques régulières. Les capillaires sont typiquement dilatés et proéminents.
La composante épithéliale est caractérisée par des kystes cornés ou par des proliférations avec des aspects trichoblastiques.
Les critères de distinction avec la mucinose cutanée focale sont une taille plus importante, un dépôt mucineux lobulé et bien démarqué qui atteint aussi le tissu sous-cutané, l’aspect proéminent et dilaté des capillaires, la composante épithéliale, les fibres réticulaires proéminentes.
Cette distinction est importante car le myxome est une vraie tumeur qui peut récidiver en cas d’excision incomplète.
De plus, le myxome peut être le signe du syndrome de Carney.







")



{kind=link}