



















La maladie périodique, ou fièvre méditerranéenne familiale, est une affection héréditaire atteignant le plus souvent les sujets originaires de l’Est méditerranéen, Juifs séfarades et Arméniens en particulier.
Son pronostic est dominé par le risque d’amylose dont la fréquence varie selon les groupes ethniques et dont la survenue conduit à l’insuffisance rénale terminale.
Le traitement continu par la colchicine, utilisé depuis 1972, permet dans la grande majorité des cas de réduire considérablement la fréquence et la gravité des accès et a permis de transformer le pronostic en prévenant le développement de l’amylose.
Caractères généraux :
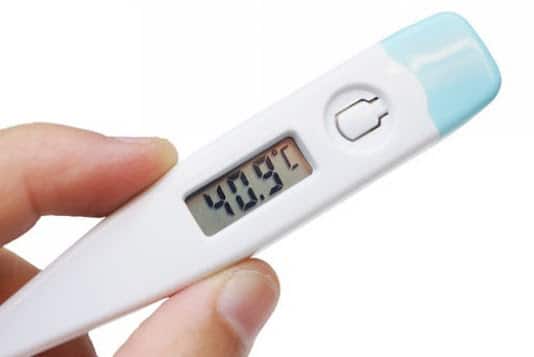 La maladie périodique est une affection chronique, caractérisée par la survenue à intervalles irréguliers de crises brèves autolimitées, associant de manière variable une fièvre persistant pendant 24 à 48 heures et des douleurs abdominales, thoraciques ou articulaires.
La maladie périodique est une affection chronique, caractérisée par la survenue à intervalles irréguliers de crises brèves autolimitées, associant de manière variable une fièvre persistant pendant 24 à 48 heures et des douleurs abdominales, thoraciques ou articulaires.
L’identification récente du gène de cette maladie (Babior BM, New Engl J Med, 1997 ; 337 : 1548-9) sur le bras court du chromosome 16 devrait permettre dans un avenir proche d’obtenir une confirmation formelle du diagnostic, fondé essentiellement jusqu’à présent sur le caractère spontanément régressif et récurrent des accès et sur la notion du terrain ethnique.
La maladie périodique touche surtout les Juifs séfarades, les Arméniens et les Arabes du Moyen-Orient.
Elle n’est cependant pas exceptionnelle chez les Juifs ashkénazes, et un certain nombre de cas ont été rapportés dans d’autres populations : turque essentiellement, mais aussi dans plusieurs pays d’Europe.
Les caractéristiques épidémiologiques de la maladie périodique suggèrent fortement l’existence d’une anomalie transmise de manière autosomique récessive, avec une pénétrance incomplète et un sex-ratio de l’ordre de trois hommes pour deux femmes.
Il n’y a pas de corrélation entre la fréquence et l’intensité des accès paroxystiques et la survenue de l’amylose, et deux expressions phénotypiques de la maladie peuvent être rencontrées : dans le phénotype I, le plus fréquent, les accès paroxystiques apparaissent les premiers, tandis que dans le phénotype II, c’est l’amylose qui survient d’abord, suivie ou non des crises paroxystiques.
Ces dernières années ont été marquées par une série de travaux ayant permis de localiser le gène de la maladie périodique, appelé MEF, sur le bras court du chromosome 16.
Symptomatologie :
La symptomatologie de la maladie périodique présente deux aspects : les manifestations paroxystiques, bruyantes mais d’évolution le plus souvent favorable spontanément, et l’amylose dont la survenue éventuelle conditionne le pronostic.
A – Manifestations paroxystiques :
Les symptômes de la maladie périodique surviennent une fois sur deux dans les 10 premières années de la vie et dans plus de huit cas sur dix avant l’âge de 20 ans.
Dans la grande majorité des cas, l’affection commence par une crise aiguë abdominale, posant le problème d’une urgence de type chirurgical.
La fréquence des différentes manifestations paroxystiques dans les séries les plus importantes, ainsi que de leurs associations.
Les accès apparaissent brusquement, atteignant leur acmé en quelques heures, et régressent habituellement en quelques jours.
Ils se répètent de manière totalement imprévisible, parfois déclenchés par certains facteurs : activité physique inhabituelle, traumatisme, émotion ou période menstruelle notamment.
1- Accès fébrile :
Il se traduit par une élévation brusque de la température qui atteint 38 à 39 °C (parfois 40 °C) en quelques heures.
Il peut être isolé, « pseudopalustre » ou accompagner une manifestation viscérale.
La fièvre s’atténue habituellement en 12 à 24 heures, mais peut persister jusqu’à 5 jours, voire plus longtemps, en particulier lorsqu’il existe une atteinte articulaire.
2- Accès péritonéal :
Il est la manifestation la plus caractéristique et, avec la fièvre, le symptôme le plus fréquent de la maladie périodique.
Il simule une urgence chirurgicale, avec parfois défense, voire véritable contracture pariétale, ou aspect radiographique évoquant une occlusion intestinale.
Lorsqu’un tel tableau est inaugural, ce qui se produit dans plus d’un cas sur deux, l’intervention chirurgicale est presque inévitable. En l’absence d’intervention, la douleur commence à régresser après 6 à 12 heures et sa disparition, complète en 24 à 48 heures, s’accompagne souvent d’une diarrhée transitoire.
Le diagnostic différentiel avec une urgence chirurgicale est souvent très délicat, reposant sur une analyse sémiologique rigoureuse et sur une surveillance soigneuse.
La prolongation de la crise pendant plus de 24 heures doit faire reconsidérer le diagnostic et renforcer la surveillance pour ne pas laisser passer l’heure d’une intervention chirurgicale.
3- Crises articulaires :
Elles touchent surtout les grosses articulations, notamment le genou, la cheville, la hanche et l’épaule.
Il s’agit habituellement d’une monoarthrite, plus rarement d’une oligoarthrite ou d’une polyarthrite. Les accès articulaires peuvent se présenter sous deux formes.
Les accès aigus, les plus fréquents, réalisent un tableau d’arthrite, avec parfois épanchement fugace constitué d’un liquide d’aspect clair, trouble ou puriforme, contenant de 200 à 1 000 000 d’éléments par mm3, polynucléaires neutrophiles non altérés essentiellement.
La crise atteint son acmé en 2 à 3 jours, puis régresse en 1 semaine environ, le plus souvent sans aucune séquelle. Les formes prolongées sont moins fréquentes et intéressent surtout le genou et la hanche.
Le tableau est celui d’une monoarthrite chronique qui s’accompagne souvent d’une attitude en flessum et d’une déminéralisation osseuse, souvent importante.
Les symptômes ne commencent à régresser qu’après un délai de plusieurs mois à 1 an et finissent par disparaître, le plus souvent sans séquelle.
Parfois néanmoins, une arthropathie destructrice chronique se développe au genou et surtout à la hanche, compromettant alors le pronostic fonctionnel.
4- Accès thoraciques :
Ils réalisent un tableau de pleurésie aiguë fébrile régressant totalement en 24 à 48 heures.
5- Signes cutanés :
Ils se traduisent surtout par un érythème érysipélatoïde siégeant aux membres inférieurs, ou par diverses autres lésions, avec parfois une vascularite, qui peuvent prendre l’aspect d’une véritable périartérite noueuse ou d’un purpura rhumatoïde.
Sont encore signalées la survenue d’une orchite aiguë unilatérale chez les garçons âgés de moins de 16 ans, et exceptionnellement d’une méningite périodique aseptique ou d’une péricardite.
6- Anomalies persistant dans l’intervalle des crises :
Une hépatomégalie, une splénomégalie, la présence d’anomalies thyroïdiennes ou du fond d’oeil sont signalées dans quelques cas.
Les néphropathies amyloïdes représentent la très grande majorité des néphropathies observées au cours de la maladie périodique.
Toutefois, un petit nombre de néphropathies non amyloïdes ont été rencontrées au cours de cette affection.
B – Amylose de la maladie périodique :
1- Caractères généraux de l’amylose :
La survenue d’une amylose au cours de la maladie périodique transforme une affection invalidante mais bénigne en une maladie mortelle quasi certaine.
Son incidence est élevée chez les Juifs originaires d’Afrique du Nord et chez les Turcs et plus faible chez les Arméniens, les Arabes et les Juifs ashkénazes.
Dans la majorité des cas décrits, l’amylose touche des sujets jeunes, souvent avant la 20e année, et vient compliquer une maladie périodique déjà connue.
La substance amyloïde de la maladie périodique est formée de fibrilles identiques à la protéine AA de l’amylose secondaire, et elle intéresse de manière diffuse la paroi de toutes les artérioles, sauf celles du système nerveux central ; cette répartition est dominée par la localisation rénale qui est la plus précoce et la plus constante.
L’apparition d’une protéinurie au cours de la maladie périodique constitue une très forte présomption d’amylose.
La confirmation histologique peut être apportée par la biopsie rénale, la biopsie rectale (positive dans 75 à 85% des cas), voire par la biopsie médullaire.
2- Manifestations cliniques liées à l’amylose :
L’amylose rénale est de loin la plus précoce et la plus fréquente des localisations viscérales de l’amylose associée à la maladie périodique, et son évolution passe par plusieurs phases successives.
La phase préclinique est définie par la latence de l’amylose.
La phase protéinurique, asymptomatique, est caractérisée par l’existence d’une protéinurie modérée ; sa durée moyenne est de 3 à 4 ans.
La phase néphrotique, caractérisée par l’apparition d’un syndrome néphrotique clinique et biologique, laisse place, après en moyenne 1 ou 2 ans d’évolution, à la phase urémique avec insuffisance rénale, qui progresse en règle rapidement, pour atteindre son stade terminal 12 à 18 mois plus tard.
Avant l’ère de l’hémodialyse, la durée de survie était en moyenne de 7 ans après l’apparition de la protéinurie et de 3 ans après celle de l’insuffisance rénale.
Les patients atteints de maladie périodique et d’insuffisance au stade terminal sont de bons candidats à l’hémodialyse et à la greffe rénale.
Le risque essentiel est alors la poursuite du processus amyloïde dans les autres organes (coeur, intestin et surrénales notamment) et sa récidive éventuelle sur le greffon, complications dont la survenue pourrait être évitée ou freinée par la poursuite du traitement par la colchicine.
Signes biologiques :
Le diagnostic de la maladie périodique reste encore purement clinique car, malgré de nombreuses recherches, aucun marqueur biologique spécifique de l’affection n’a pu être mis en évidence.
De nombreuses anomalies biologiques ont néanmoins été signalées, dont les plus courantes sont l’hyperleucocytose et l’augmentation de la vitesse de sédimentation globulaire, correspondant à une augmentation du fibrinogène et des alpha2-globulines.
L’enquête immunologique, et notamment la recherche d’autoanticorps, est habituellement négative.
Évolution :
Après une période de latence, qui dans la majorité des cas est brève et ne dépasse pas l’enfance, la maladie est caractérisée par la survenue d’accès aigus séparés par des périodes asymptomatiques de durée irrégulière, variant de quelques jours à plusieurs années.
L’effet bénéfique de la grossesse sur le cours évolutif de la maladie périodique est signalé par la majorité des auteurs, avec une diminution de la fréquence et de la sévérité des accès, voire une rémission complète, jusqu’à l’accouchement, qui peut se poursuivre pendant la lactation.
Le pronostic de la maladie périodique dépend essentiellement du risque d’amylose et a été complètement modifié par l’efficacité préventive de la colchicine.
Si l’amylose n’apparaît pas, le pronostic est relativement bon, et pour certains auteurs, la durée de survie serait identique à celle des sujets normaux.
Critères du diagnostic :
En l’absence de marqueur biologique spécifique, le diagnostic de la maladie périodique repose sur une analyse sémiologique rigoureuse, confirmée éventuellement par un test thérapeutique par la colchicine.
Dans un proche avenir, l’identification du gène de la maladie périodique devrait permettre de porter le diagnostic de certitude dans les cas difficiles.
Traitement :
Depuis 1972, l’immense majorité des cas de maladie périodique est traitée de manière continue par la colchicine.
La posologie habituelle est de l’ordre de 1 mg/j en 1 ou 2 prises, quels que soient le poids et l’âge.
Cette méthode thérapeutique permet, dans la grande majorité des cas, de faire disparaître totalement, ou tout au moins d’espacer, les accès.
Tous les auteurs signalent cependant un certain taux d’échecs, le plus souvent proche de 10 %, même lorsque le traitement est scrupuleusement suivi.
L’efficacité curative de la colchicine sur une crise déjà installée est beaucoup plus inconstante, même en utilisant des doses plus élevées.
L’utilisation prolongée de la colchicine est susceptible d’entraîner des effets indésirables tels qu’une diarrhée et des douleurs abdominales, qui régressent à la diminution de la posologie et n’entravent que rarement la poursuite du traitement.
Les craintes majeures concernent les éventuels risques sur la reproduction que pourrait induire l’utilisation continue d’un tel médicament chez une patiente en période d’activité génitale et en âge de procréer ; ces risques paraissent cependant limités, et la naissance d’enfants normaux a été observée à plusieurs reprises alors que l’un ou l’autre des parents avait poursuivi son traitement sans interruption au moment de la fécondation ou pendant la grossesse.
Pour les couples projetant une grossesse alors qu’un des deux conjoints est traité par la colchicine, les médecins de l’université de Tel-Aviv, s’appuyant sur une expérience importante, proposent de poursuivre le traitement par la colchicine durant la grossesse et de procéder systématiquement à une amniocentèse afin de réaliser un avortement si celle-ci révélait des anomalies chromosomiques foetales, ce qui n’a été le cas que de manière tout à fait exceptionnelle chez leurs patientes.
Il est maintenant clairement établi que le traitement continu par la colchicine prévient, ou tout au moins retarde de manière significative, l’apparition de l’amylose dans l’immense majorité des cas, même lorsqu’il reste sans effet sur la répétition des accès paroxystiques.
L’action curative de la colchicine vis-à-vis d’une amylose déjà déclarée est moins démonstrative mais peut se traduire par la stabilisation, la régression et même parfois la disparition de la protéinurie, à condition que celle-ci ne soit pas trop évoluée et que des posologies supérieures à 1,5 mg/j soient utilisées.
Ces résultats justifient les recommandations des auteurs israéliens qui conseillent de soumettre systématiquement à un traitement continu par la colchicine, à une dose quotidienne comprise entre 1 à 2 mg, tout malade atteint de maladie périodique, et ce dès que le diagnostic de l’affection a été porté avec certitude.
Ce traitement doit être poursuivi même s’il demeure inefficace sur les accès paroxystiques, notamment chez les malades en hémodialyse ou ayant eu une transplantation rénale, pour prévenir ou retarder le développement de l’amylose dans d’autres organes que les reins, ainsi que dans le transplant rénal.







")



{kind=link}