



















Introduction :
La maladie de Darier a été décrite de façon indépendante en 1889 par Darier, sous le nom de psorospermose folliculaire végétante et par White, sous celui de kératose folliculaire.
C’est une génodermatose rare, transmise en dominance autosomique, associant des lésions papulokératosiques prédominant sur les zones séborrhéiques et une atteinte acrale avec des lésions unguéales caractéristiques.
Ces dernières années, les progrès en génétique et en biologie moléculaire sont à l’origine d’importantes avancées, grâce à la découverte du gène ATP2A2 sur le chromosome 12, puis de son implication dans le mécanisme pathogénique, faisant intervenir une pompe à calcium adénosine triphosphate (ATP)ase dépendante.
Ces travaux ont aussi permis d’expliquer les formes localisées, surtout segmentaires, par la théorie du mosaïcisme cellulaire.
Épidémiologie :
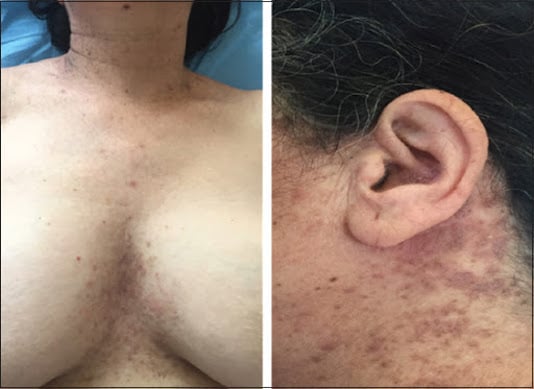 La maladie de Darier est une génodermatose peu fréquente avec un sex-ratio proche de 1.
La maladie de Darier est une génodermatose peu fréquente avec un sex-ratio proche de 1.
Sa prévalence est variable : de 1/100 000 en Scandinavie à 1/36 000 dans le nord-est de l’Angleterre.
L’incidence est estimée à quatre nouveaux cas par million d’habitants tous les dix ans.
Les premières manifestations apparaissent entre 6 et 20 ans, avec un pic à la puberté pouvant être en rapport avec des modifications de l’excrétion sébacée ou sudorale et de la flore cutanée, mais la maladie peut se révéler à tout âge.
Les formes sporadiques sont rapportées dans 29 % des cas chez les 163 patients de Burge.
Une étude récente retrouve 14 % de mutations spontanées dans la population écossaise.
Clinique :
A – FORME CLASSIQUE :
La maladie de Darier se présente de prime abord sous la forme de nappes brunâtres à contours irréguliers, émiettés, à surface rugueuse et sèche.
C’est à la périphérie que l’on retrouve la lésion élémentaire : la papule cornée caractéristique.
La lésion élémentaire est en effet une papule kératosique de 1 à 3 mm de diamètre, qui débute par une petite élevure punctiforme de couleur peau normale, puis se recouvre d’une croûte brunâtre ou grisâtre, de siège non exclusivement folliculaire.
Son arrachage laisse une petite dépression infundibuliforme.
Leur confluence aboutit à la formation de vastes placards papulokératosiques et croûteux souvent prurigineux (88 %), donnant à la peau un aspect sale, « crasseux » et rugueux à la palpation.
L’odeur désagréable, secondaire à la macération et à la prolifération bactérienne, est à l’origine d’un handicap social parfois important (44 %).
Il existe aussi des taches pigmentées lenticulaires jaunes brunâtres non kératosiques bien limitées siégeant en périphérie des plaques, et pouvant aboutir par confluence à une mélanodermie diffuse.
Elles semblent indépendantes des formations kératosiques mais, dans certains cas, elles sont aussi une forme évolutive (précoce ou tardive) des lésions kératosiques.
Les lésions prédominent au niveau des régions séborrhéiques (92 %), médiothoraciques (87 %) et médiodorsales (85 %), la lisière du cuir chevelu, particulièrement le front (84 %), les faces latérales du cou et la région sus-claviculaire (82 %), les tempes, la région rétroauriculaire et les oreilles (58 %).
L’atteinte des grands plis (axillaires, inguinaux, sous-mammaires, région anogénitale) est fréquente, mais modérée dans huit cas sur dix ; à l’opposé, les formes sévères avec lésions végétantes, papillomateuses et suintantes, sont rencontrées dans 6 % des cas.
Les lésions des extrémités sont très fréquentes (96 %) et peuvent être la première manifestation de la maladie, notamment chez l’enfant.
Dans d’autres cas, elles en sont l’unique expression et réalisent alors la maladie de Darier acrale, mais cette forme peut secondairement se compléter.
Trois localisations sont évocatrices :
– sur le dos des mains et plus rarement des pieds, on observe des lésions papuleuses, gris jaunâtre ou de la couleur de la peau normale, parfois kératosiques comparables à des verrues planes.
Ces papules kératosiques (48 %) peuvent évoquer également une épidermodysplasie verruciforme de Lewandowsky et Lutz, un syndrome de Cowden ou, le plus souvent, une acrokératose verruciforme de Hopf.
Dans ce dernier cas, l’histologie rectifie le diagnostic en ne retrouvant ni dyskératose ni acantholyse, mais une hyperkératose orthokératosique circonscrite en « clocher d’église ».
Ces lésions sont asymptomatiques, rarement prurigineuses et plus exceptionnellement douloureuses.
Il ne semble pas exister d’aggravation au soleil ;
– sur les paumes et les plantes, il existe très souvent de minuscules puits (pits) avec aspect d’hyperkératose ponctuée (84 %) ;
– sur les ongles (92 %), l’atteinte est caractéristique, réalisant une alternance de bandes longitudinales rouges sombres et blanches de la lunule jusqu’au bord libre de l’ongle.
Les ongles sont fragiles, ont un aspect strié (onychorrexhie) et présentent une encoche distale en V (onychoschizie) ainsi qu’une hyperkératose sous-unguéale cunéiforme occasionnant une gêne à l’habillage.
Plus rarement, il a été décrit des hyperkératoses périunguéales, voire des périonyxis inflammatoires.
L’atteinte unguéale est localisée en général à deux ou trois ongles, touchant plus les doigts que les orteils.
De façon anecdotique, on a rapporté des modifications des dermatoglyphes, deux cas d’hyperkératose palmoplantaire filiforme ou des kératodermies plantaires diffuses ou circonscrites.
Des macules hémorragiques palmaires sont retrouvées dans 6 % des cas de la série de Burge ; exceptionnellement, ces lésions ont été la seule expression de la maladie au sein d’une famille consanguine.
B – FORMES CLINIQUES :
Toutes les formes cliniques peuvent s’observer, l’expression phénotypique est variable, allant de l’atteinte isolée des ongles à la forme floride de la maladie.
Schématiquement, les lésions sont discrètes dans 26 % des cas, modérées dans 65 % des cas et sévères dans 9 % des cas.
1- Formes topographiques :
En dehors de la forme acrale, certaines localisations donnent un aspect plus particulier.
* Muqueuses :
L’atteinte des muqueuses est présente dans 13 à 50 % des cas.
Toutes les muqueuses malpighiennes peuvent être touchées : buccales, pharyngées, laryngées, oesophagiennes, bronchiques, anogénitales et rectales.
L’atteinte buccale est la plus fréquente, prédominant sur les gencives, la face interne des joues, le palais membraneux plus que le palais osseux.
Il s’agit d’efflorescences blanchâtres punctiformes ou lenticulaires, mais parfois maculopapuleuses érythémateuses et ombiliquées.
Elles sont tantôt isolées, tantôt coalescentes en nappes d’aspect pavimenteux.
La langue peut être villeuse ou fissurée ou blanche parsemée de points rouges.
L’atteinte peut se localiser également au niveau des glandes et des canaux salivaires, responsables d’une hypersialorrhée ou d’une xérostomie, et parfois de lithiase sous-maxillaire ou parotidienne (8 %).
La localisation aux muqueuses anogénitales est exceptionnelle, sous forme de papules rosées molles.
* Cuir chevelu :
L’atteinte est fréquente (69 %), et se caractérise par des placards squamocroûteux d’où émergent des cheveux intacts en bouquets ou en poils de brosse.
Elle peut se limiter à de simples petites élevures cornées spinulosiques, ou au contraire être très étendue à surface vermoulue et plus rarement cérébriforme.
L’alopécie reste exceptionnelle et doit faire rechercher une surinfection bactérienne.
2- Formes atypiques :
Des formes inhabituelles de la maladie ont été rapportées de manière anecdotique dans la littérature :
– les formes débutantes, parfois trompeuses, sont marquées par une simple modification de couleur et de texture de la peau qui apparaît sale et rugueuse, par de petites élevures lichénoïdes peu kératosiques ou à type de verrues planes ;
– les formes vésiculobulleuses, voire pustulobulleuses sont rares ; les bulles de petites tailles surviennent le plus souvent sur certaines plaques, mais peuvent se localiser en peau saine.
Ces lésions bulleuses sont parfois prédominantes, et leur rupture réalise des placards érodés suintants, croûteux à bordure circinée.
Elles prédominent sur le cou et les grands plis, pouvant simuler une maladie de Hailey-Hailey.
Les immunofluorescences directe et indirecte sont négatives, et ces bulles sont l’expression majeure des fentes acantholytiques.
La macération, l’exposition solaire, la prise de rétinoïdes, un geste chirurgical sont des facteurs aggravants.
L’important est d’éliminer avant tout une surinfection virale, notamment herpétique.
– les formes dyschromiques affectent principalement les mélanodermes. Associées à une atteinte classique, ces maculopapules leucodermiques, hypopigmentées, prédominent au niveau du tronc et à la racine des membres chez l’enfant ou l’adolescent.
Initialement considérées comme un phénomène postinflammatoire, ces lésions sont en fait des formes frustes, précoces de la maladie, comme le confirme l’histologie avec acantholyse dans les zones d’hypomélanose ;
– les formes hyperkératosiques sous forme de plaques verruqueuses, exophytiques siègent sur les faces d’extension des membres dans cinq observations.
L’atteinte isolée des mamelons et de la région aréolaire est également possible ;
– les formes hypertrophiques malodorantes s’observent avant tout dans les grands plis et sont favorisées par la macération ;
– les formes comédoniennes, essentiellement localisées à la face et sur le cuir chevelu, s’associent à des pits palmaires et à une onychopathie.
Les lésions ne sont pas aggravées par l’exposition solaire, et siègent également en zone non photoexposée.
Elles sont à différencier d’une dyskératose acantholytique comédonienne.
3- Formes localisées et segmentaires :
La maladie de Darier peut être localisée à une seule région tégumentaire, ou plus curieusement systématisée en bande ou zoniforme.
Ces formes segmentaires sont rares, et représentent environ 10 % des formes de la maladie.
L’âge de survenue est plus tardif, aux environs de 30-40 ans, sans prédominance de sexe.
L’atteinte cutanée est de sévérité variable, et se traduit par des lésions papulokératosiques unilatérales blaschkolinéaires.
Cependant, dans certains cas, il existe une atteinte unguéale, des pits palmoplantaires, des lésions papuleuses du dos des mains, voire une atteinte des muqueuses confortant son rattachement à la maladie de Darier.
Ces lésions sont parfois aggravées par les ultraviolets B (UVB) ou la chaleur. Cette forme systématisée linéaire est appelée par Happle forme segmentaire de type 1.
Elle s’expliquerait par une mutation postzygotique survenant précocement lors de l’embryogenèse.
La mutation postzygotique n’est pas transmissible, sauf s’il s’associe un mosaïcisme germinal.
Mais à ce jour, il n’existe pas d’observation de descendant de patient atteint de forme segmentaire ayant une forme classique de maladie de Darier.
De manière anecdotique, on a rapporté l’association à une agénésie rénale controlatérale et à un syndrome de Gardner.
Plus intrigant sur le plan clinique sont cinq cas de maladie de Darier associant des lésions diffuses avec des zones de renforcement segmentaire.
Cette forme trouve son explication dans la théorie du mosaïcisme qu’Happle individualise sous le nom de forme segmentaire de type 2.
Le principal diagnostic différentiel, mais qui pose un problème nosologique, est le nævus acantholytique et dyskératosique à disposition linéaire, dont l’aspect clinique et histologique est identique à la maladie de Darier segmentaire.
Happle a été le premier à suggérer que cet hamartome serait une forme segmentaire de la maladie de Darier.
Cette hypothèse a été confortée par la découverte de la mutation du gène ATP2A2 dans les nævus acantholytique et dyskératosique blaschkolinéaires.
La mutation n’a été détectée qu’en peau lésée.
Elle est absente en peau saine et dans les leucocytes circulants.
Aussi, cette découverte confirmerait que le nævus acantholytique dyskératosique ne serait qu’une forme localisée segmentaire de la maladie de Darier.
Histologie :
Bien que très évocateur, l’aspect histologique n’est absolument pas spécifique.
A – MICROSCOPIE OPTIQUE :
On observe une acantholyse avec formation de fentes suprabasales qui s’étendent de façon irrégulière dans le stratum spinosum.
À cette acantholyse s’associe une kératinisation précoce et anormale, aboutissant à la formation de cellules dyskératosiques dans le corps muqueux (corps ronds) et dans le stratum corneum (grains).
Les corps ronds sont de gros kératinocytes dont le noyau est hyperbasophile, irrégulier, parfois pycnotique entouré d’un cytoplasme fortement éosinophile et vacuolisé.
Ils siègent dans la partie supérieure du corps muqueux, dans la couche granuleuse et au sein des fentes.
Les grains sont de petits kératinocytes au cytoplasme peu abondant, en général sans noyau.
Ils sont présents au sein des fentes et dans la couche cornée.
On observe également une papillomatose, et une hyperkératose associée à des bouchons de kératine au sein des follicules pilosébacés.
Cependant, l’atteinte palmoplantaire ou des muqueuses prouve que l’affection n’est pas exclusivement folliculaire.
Les lésions vésiculobulleuses correspondent à une accentuation du phénomène d’acantholyse.
Aux niveaux acral et muqueux, le clivage et la dyskératose sont parfois discrets, nécessitant des coupes sériées pour être mis en évidence.
Les formes hypertrophiques réalisent une prolifération pseudoépithéliomateuse s’enfonçant dans le derme.
Les fentes suprabasales sont remplies d’hématies dans les formes hémorragiques.
B – MICROSCOPIE ÉLECTRONIQUE :
L’acantholyse résulte de la perte de la cohésion interkératinocytaire.
La microscopie électronique met en évidence une atteinte du système desmosome-tonofilaments avec raréfaction des desmosomes qui sont rudimentaires et isolés des tonofilaments.
Le contact entre les kératinocytes s’établit surtout par des replis des membranes cytoplasmiques avec présence de fentes interkératinocytaires.
La dyskératose correspond à une agglutination des tonofilaments autour du noyau, associée à des grains de kératohyaline dans la couche granuleuse.
L’acantholyse précède la dyskératose, l’anomalie primitive se situant au niveau du complexe desmosome-tonofilaments.
C – IMMUNOHISTOCHIMIE :
Il existe un marquage cytoplasmique diffus des protéines de la plaque desmosomale (desmoplakine 1, 2 et plakoglobine) au sein des cellules acantholytiques.
Les jonctions d’adhérence (vinculine) et les jonctions gap (connexine) apparaissent préservées.
Les cadhérines desmosomales (desmogléine et desmocolline) sont également exprimées dans le cytoplasme de la plupart des cellules acantholytiques.
L’aspect du marquage semble dépendre du stade de l’acantholyse ; il est granuleux quand les lésions sont précoces, puis diffus à un stade plus tardif.
En affinant la technique, Hakuno et Kowalewski ont récemment démontré que le marquage concernait en fait les épitopes intracellulaires des cadhérines desmosomales (desmogléine 1, 3, desmocolline).
Hakuno a également mis en évidence une atteinte des épitopes extracellulaires de la E-cadhérine des jonctions d’adhérence.
La dissociation des domaines intra- et extracellulaires des cadhérines desmosomales et classiques (E-cadhérine) serait caractéristique de l’acantholyse dans la maladie de Darier et dans le pemphigus bénin familial.
Elle n’est pas retrouvée dans le pemphigus vulgaire.
Récemment, on a observé que la P-cadhérine, protéine majeure des jonctions d’adhérence, était exprimée à la surface des kératinocytes basaux et suprabasaux en peau périlésionnelle, alors qu’elle était uniquement localisée à la couche basale en peau non lésionelle.
Cette surexpression de la P-cadhérine serait liée à des variations de flux calciques.
Maladies associées :
Les maladies associées sont relativement rares et dominées par les troubles neurologiques : épilepsie, retard mental, encéphalopathie, atrophie cérébrale et manifestations psychiatriques (troubles de l’humeur, dépression, schizophrénie, psychose maniacodépressive et suicide).
Le handicap causé par la maladie est un facteur supplémentaire de repli sur soi et d’isolement.
Les études génétiques n’ont pas encore mis en évidence de profil mutationnel spécifique, mais cela reste très discuté.
On a également rapporté de manière non spécifique des anomalies génito-urinaires (agénésie rénale et testiculaire), oculaires (lésions cornéennes, rétinites pigmentaires), deux cas de pelvispondylite rhumatismale, des taches café-au-lait sans autre élément pour une neurofibromatose, des anomalies osseuses (déminéralisation, kystes osseux diaphysaires) et de multiples kystes épidermoïdes.
Il ne semble pas exister de prédisposition particulière aux tumeurs, bien que l’on ait observé des carcinomes sur des lésions cutanées de Darier (carcinome basocellulaire ou épidermoïde) ou muqueuses (cancer de l’oesophage).
L’association à un dermatofibrosarcome est probablement fortuite.
Évolution :
Dans la majorité des cas, l’évolution est chronique, alternant des périodes de stabilisation et des poussées spontanées ou provoquées.
Cependant dans deux cas sur trois, la maladie s’améliore avec l’âge ou demeure stationnaire ; à l’inverse, on assiste plus rarement à une aggravation progressive des lésions, mais le pronostic vital reste favorable.
Différents facteurs peuvent favoriser les poussées.
A – STRESS (43 %), CHALEUR, SUDATION ET SOLEIL :
La photosensibilité est fréquente (un cas sur deux dans la série de Burge) mais le caractère photodéclenché est plus rare.
L’apparition de lésions en peau saine après exposition à doses infraérythémales en UVB plaide en faveur d’une action directe du rayonnement sur la dermatose, car elles surviennent en dehors de tout érythème actinique.
À doses supraérythémales (10 doses érythémateuses minimales [DEM]), l’induction des lésions est en rapport avec un phénomène de Koebner.
En revanche, les UVA n’ont aucune influence. De façon paradoxale, le soleil peut améliorer les lésions dans 6 % des cas.
B – MÉDICAMENTS :
Des poussées sont parfois observées après une anesthésie générale ou la prise de lithium.
Pour ce dernier, le mécanisme est incertain, mais la diminution d’acide adénosine monophosphorique (AMP) cyclique kératinocytaire, induite par le lithium, entraîne une prolifération épidermique.
Des précautions d’emploi sont donc nécessaires, en cas d’association de maladie de Darier à une psychose maniacodépressive.
C – AUTRES :
Les règles, la grossesse et la ménopause ne semblent pas être des facteurs aggravants.
Dans certains cas, la prise d’oestroprogestatifs permettrait le contrôle des poussées déclenchées par les cycles.
Complications :
Le retentissement psychologique est variable ; le handicap social est surtout marqué à l’adolescence (63 %) en raison du caractère parfois affichant de la maladie, et de la gêne secondaire à la macération (44 %).
À l’âge adulte, l’activité professionnelle n’est perturbée que dans 16 % des cas.
Les complications sont principalement infectieuses, et sont suspectées en cas d’aggravation brutale et douloureuse des lésions.
Il existe une sensibilité particulière aux infections virales, notamment herpétiques (14 %), avec apparition d’un tableau de pustulose varioliforme de Kaposi-Juliusberg parfois généralisé.
Des infections à coxsackie A16, réalisant le même tableau clinique, ont également été rapportées.
Les surinfections mycosiques trichophytiques et bactériennes sont possibles ; la colonisation des lésions kératosiques est en fait fréquente.
La persistance intrakératinocytaire de Staphylococcus aureus est à l’origine d’infections répétées et d’échecs thérapeutiques aux antibiotiques.
Cette susceptibilité aux infections résulterait plus de la perte de l’effet barrière de la peau, que d’une réelle anomalie de l’immunité.
En effet, il n’existe pas d’altération immunitaire constante et significative, bien que certains auteurs aient rapporté un déficit de l’immunité cellulaire.
Ces poussées inflammatoires d’origine infectieuse sont à distinguer d’une eczématisation ou d’une irritation secondaires à l’application de topiques, notamment antiseptiques.
Diagnostic différentiel :
Chez l’adolescent, les formes modérées sont parfois confondues avec une acné ou une dermite séborrhéique, mais les principaux diagnostics différentiels sont la maladie de Hailey-Hailey, la maladie de Grover et, à un moindre degré, la maladie de Galli-Galli.
A – MALADIE DE HAILEY-HAILEY (PEMPHIGUS CHRONIQUE BÉNIN FAMILIAL) :
Cette dermatose acantholytique héréditaire à transmission autosomique dominante est due à une mutation sur un locus du chromosome 3 en position q21-24 ; le gène ATP2C1 codant pour une pompe à calcium ATPase dépendante a été récemment identifié.
La maladie débute à l’adolescence ou à l’âge adulte, et les lésions prédominent dans les plis et la région cervicale.
Dans certains cas, l’atteinte peut être plus diffuse et affecter le tronc, les membres et le cuir chevelu.
L’aspect clinique initial est celui de lésions vésiculobulleuses dont la rupture aboutit à la formation de plaques érosives et suintantes.
Au niveau inguinal, ces placards sont parcourus de fissures en rhagades parallèles très caractéristiques.
L’atteinte muqueuse est possible. Des bandes blanches isolées sans fragilité unguéale sont présentes dans 70 % des cas.
L’influence saisonnière est de règle et le handicap social parfois marqué, comparables à la maladie de Darier.
Sur le plan histologique, l’acantholyse prédomine sur la dyskératose.
L’immunofluorescence directe est négative.
B – MALADIE DE GROVER (DERMATOSE ACANTHOLYTIQUE TRANSITOIRE OU PERSISTANTE) :
Elle débute plus tardivement et touche classiquement l’homme de plus de 40 ans, généralement pendant la période hivernale.
L’éruption est faite de papules érythémateuses ou violacées, ou de papulovésicules isolées ou regroupées en petites plaques.
Elles prennent souvent l’aspect de kératose séborrhéique ou actinique irritée.
Le prurit est fréquent et les lésions siègent dans la région dorsale, lombaire ou thoracique.
La chaleur, la sudation, la fièvre, l’alitement, la période postopératoire sont des facteurs aggravants.
Dans la plupart des cas, la maladie est transitoire, mais des récurrences sont possibles.
La forme persistante est considérée par certains comme une forme fruste de maladie de Darier, mais aucune mutation du gène ATP2A2 n’a été mise en évidence.
Sur le plan histologique, l’acantholyse peut reproduire l’image d’une maladie de Darier ou Hailey-Hailey.
L’immunofluorescence directe est négative.
C – MALADIE DE GALLI-GALLI :
C’est une génodermatose rare qui se présente sous forme de nappes pigmentaires réticulées parsemées d’éléments papulokératosiques brunâtres, localisée au niveau des grands plis, du cou, des faces latérales du tronc et du dos des mains.
Cet aspect clinique peut être trompeur d’autant qu’en histologie, il existe des fentes acantholytiques.
Cependant, la localisation privilégiée au niveau des grands plis, l’aspect réticulé et l’absence de dyskératose à l’histologie permettent d’écarter facilement la maladie de Darier.
Cette affection peu connue est considérée comme la variante acantholytique de la maladie de Dowling-Degos.
D – AUTRES :
Pour les formes topographiques, le dyskératome verruqueux solitaire, bien qu’ayant une image histologique similaire à celle de la maladie de Darier, reste une entité bien distincte.
Les formes localisées aux grands plis font discuter l’exceptionnel intertrigo acantholytique dyskératosique, dont l’authenticité reste discutée.
Génétique :
A – CHROMOSOME :
Au cours de la dernière décennie, le gène responsable a été localisé sur le chromosome 12 en position q 23-24,1 sans évidence pour une hétérogénéité de ce locus.
B – GÈNE ATP2A2 :
Le gène ATP2A2, composé de 21 exons, de 20 introns et de deux sites d’épissage alternatifs s’étend sur 76 kb approximativement.
Il code pour une protéine appelée SERCA2 (sarcoendoréticulum calcium), fortement exprimée au niveau kératinocytaire.
C – PROTÉINE SERCA2 :
1- Structure :
La protéine SERCA2 est une pompe à calcium localisée sur la paroi du réticulum endoplasmique et sur l’appareil de Golgi des kératinocytes et des fibroblastes.
Cette pompe à calcium présente cinq domaines fonctionnels, dont quatre sont cytoplasmiques (un domaine de liaison du calcium, un domaine de phosphorylation, un domaine de fixation ou de liaison de l’ATP, un domaine charnière) et un domaine transmembranaire.
2- Rôle :
SERCA2 agit comme une pompe à calcium ATPase dépendante.
Le calcium est localisé dans les compartiments extracellulaire (le cytosol) et intracellulaire (le réticulum endoplasmique).
Le cytosol ayant habituellement une faible concentration de calcium, les échanges calciques se font du réticulum vers le cytosol selon un gradient de concentration.
Ce flux de calcium est dénommé signal calcique.
Une fois le signal terminé, le retour du calcium dans le réticulum endoplasmique se fait par l’intermédiaire d’une pompe à calcium ATPase dépendante comme SERCA2.
L’énergie nécessaire au transport du calcium est libérée par hydrolyse de l’ATP.
D – MUTATIONS DU GÈNE ATP2A2 :
Plus de 70 mutations ont été décrites au sein de 47 familles européennes différentes ou dans des cas sporadiques.
Elles s’étendent tout le long du gène ATP2A2 et sont responsables d’altérations du fonctionnement des principaux domaines de la pompe à calcium.
Selon Ruiz-Perez, il existe une majorité de mutations non-sens et faux-sens.
In vitro, ces altérations se manifestent par des dysfonctions des sites de fixation calcique et des anomalies de phosphorylation.
Ces mutations sont responsables le plus souvent de la synthèse d’une protéine tronquée ayant perdu son activité normale.
Un seul allèle ne suffit pas pour répondre aux besoins cellulaires.
Ainsi, une diminution d’environ 50 % du produit du gène entraîne des manifestations au niveau phénotypique.
Les effets de ces mutations sont donc compatibles avec le principe d’haplo-insuffisance, car seul l’allèle sauvage code pour une protéine normale, mais en quantité insuffisante.
Ce fait explique probablement le mode autosomique dominant de transmission de la maladie.
En général, il ne semble pas exister de lien entre le profil mutationnel et l’expression phénotypique de la maladie, sauf pour certaines formes cutanées atypiques notamment hémorragiques, qui seraient associées aux mutations faux-sens.
Dans ce cas, les mutations seraient responsables, au sein des cellules endothéliales, de l’interruption de la fonction de SERCA2, et/ou de la synthèse de protéines mutantes ayant une action vasculaire.
E – GÉNÉTIQUE ET FORMES PSYCHIATRIQUES :
Pour les formes psychiatriques ou neuropsychiatriques, certains suspectent des mutations faux-sens surtout localisées sur la région 3’ du gène, mais cela est très contesté. Jacobsen, notamment, ne retient pas le gène de la maladie de Darier comme un gène de susceptibilité de la maladie bipolaire.
F – APPROCHE PHYSIOPATHOLOGIQUE : RELATION ENTRE PROTÉINE ANORMALE ET PHÉNOTYPE CUTANÉ
Les observations réalisées lors d’études immuno-histochimiques et en microscopie électronique montrent que des anomalies qualitatives ou structurales des protéines desmosomales seraient impliquées dans les phénomènes d’acantholyse et les anomalies de la kératinisation.
L’identification récente du gène ATP2A2 suggère la participation du signal calcique dans les mécanismes d’adhésion kératinocytaire et dans la formation des desmosomes.
In vitro, les cellules épithéliales cultivées en présence d’anticorps inhibiteurs sélectifs des pompes à calcium ont des anomalies d’adhésion cellulaire.
Plus précisément, on a pu mettre en évidence un retard du transport des desmoplakines vers la membrane cytoplasmique, et une altération des fonctions de régulation de phosphorylation ou de déphosphorylation de la desmoplakine.
Des travaux confirment le rôle du signal calcique dans la sélection des composants de la plaque desmosomale, dans l’assemblage des protéines ou dans l’ancrage des tonofilaments.
Traitement :
Il n’existe actuellement pas de traitement curatif.
Les mesures, uniquement suspensives, visent à améliorer la tolérance fonctionnelle tout en favorisant l’insertion socioprofessionnelle dans les formes graves.
Des recommandations simples comme l’éviction solaire ou la photoprotection, le port de vêtements amples, doux en coton, l’usage de crèmes émollientes, la lutte contre la transpiration ont une action préventive contre les poussées.
L’utilisation de faux ongles limite la gêne à l’habillage.
Le patient doit bénéficier d’une information claire sur l’évolution et les complications possibles de sa maladie.
Un conseil génétique peut être utile dans certains cas, afin d’évaluer le risque de transmission.
A – TRAITEMENTS LOCAUX :
1- Traitements symptomatiques :
Les crèmes émollientes plus ou moins kératolytiques (urée, acide lactique) améliorent la trophicité cutanée et soulagent le prurit.
Les antibiotiques topiques peuvent lutter ponctuellement contre les surinfections.
Les corticoïdes locaux, utilisés sur de courtes durées, peuvent contrôler une eczématisation.
2- Traitements spécifiques :
Les rétinoïdes topiques : l’isotrétinoïne gel à la concentration de 0,05 % a été efficace en 3 mois en application biquotidienne, au prix d’une irritation locale parfois importante.
Des concentrations supérieures à 0,1 % sont mal tolérées et sans bénéfice supplémentaire.
Le tazarotène gel à 0,05 % (Zoract) associé à un corticoïde topique améliorait les lésions en 6 semaines.
Des résultats identiques sans dermocorticoïdes étaient obtenus à la concentration de 0,01 %.
L’adapalène gel à 0,1 % (Differinet) utilisé de façon ponctuelle a donné de bons résultats.
Les émollients améliorent la tolérance cutanée des rétinoïdes qui sont déconseillés pendant la grossesse.
Le 5-fluoro-uracile à 1 % (Efudixt) a été efficace chez deux patients sous rétinoïdes oraux.
La guérison des lésions était obtenue en 15 jours avec maintien à 6 mois.
La résorption cutanée est faible (6 %) sans effet systémique notable.
Les essais sont contradictoires avec le calcipotriol (Daivonex), efficace pour certains, très irritant pour d’autres.
Tous ces résultats méritent en fait d’être confirmés sur des séries contrôlées.
B – TRAITEMENTS GÉNÉRAUX :
La vitamine A seule (50 000 UI/j) ou en association aux traitements locaux, a longtemps été le seul traitement proposé et d’efficacité variable, mais elle exposait aux risques d’hypervitaminose A.
Les rétinoïdes constituent le traitement de référence.
Ils régulent la prolifération cellulaire, favorisent la différenciation épidermique et modulent l’expression des gènes kératinocytaires.
L’acitrétine (Soriatanet) est prescrit à la dose initiale de 0,25 à 0,5 mg/kg/j.
L’isotrétinoïne (Roaccutanet) utilisée à la dose de 0,5 à 1 mg/kg/j est moins contraignante chez la femme en période d’activité génitale.
Ces deux molécules semblent avoir une efficacité comparable, mais il n’existe actuellement pas d’étude comparative.
Le bénéfice est maximal au bout de 2 à 3 mois, mais la fréquence des effets secondaires, souvent dose-dépendante, peut être un facteur limitant.
Ainsi, à moyen terme seulement, 60 % des patients poursuivent leur traitement.
Le but est donc d’obtenir une dose minimale efficace, généralement comprise entre 10 et 25 mg d’acitrétine.
Les traitements séquentiels améliorent particulièrement la tolérance pendant la période estivale.
L’étrétinate a été proposé en cas d’échec de l’acitrétine mais il n’est plus disponible en France.
La ciclosporine (3 à 5 mg/kg/j) a des résultats contradictoires et, malheureusement, ces traitements sont uniquement suspensifs. L’efficacité des acides gras essentiels mérite d’être confirmée.
C – TRAITEMENTS CHIRURGICAUX :
Ce sont les seuls traitements radicaux qui exposent cependant au risque de Koebner ou de récidive à plus long terme.
L’exérèse chirurgicale avec suture directe ou greffe de peau mince est possible dans les formes limitées.
La dermabrasion est efficace dans les lésions hypertrophiques des plis ; en région thoracique, elle expose au risque de chéloïde.
La vaporisation au laser CO2 entraîne une cicatrisation lente ; le laser Erbium-YAG, moins traumatisant, est indiqué dans le traitement des lésions récalcitrantes.
D – INDICATIONS :
Dans les formes modérées, seuls les traitements locaux (émollients, kératolytiques, antibiotiques, voire rétinoïdes topiques) sont utilisés.
Dans les formes étendues ou sévères, le traitement repose sur les rétinoïdes de synthèse, essentiellement l’acitrétine.
Leur utilisation est difficile en cas de lésions érosives ou bulleuses, car ils peuvent accroître la fragilité cutanée. Les formes hypertrophiques des plis répondent mal aux rétinoïdes et la chirurgie peut être nécessaire.
En cas de complication, la corticothérapie orale à faible dose (20 à 40 mg/j) peut être indiquée dans les formes eczématisées étendues et vésiculobulleuses après avoir éliminé une surinfection.
Une antibiothérapie générale adaptée est prescrite en cas de surinfection bactérienne ; les surinfections herpétiques seront traitées par aciclovir.

. Traitement (Traitements anti-« tumor necrosis factor » (TNF) dans la polyarthrite rhumatoïde)")





")


")
{kind=link}