



















Introduction :
Le terme de livedo dérive du mot latin « lividus » correspondant à des taches bleues.
Dans le langage médical, il est utilisé depuis Hébra et Kaposi pour désigner des marbrures violacées dessinant un réseau d’origine vasculaire.
Il s’est progressivement imposé d’où l’abandon de ses anciens synonymes tels que « inflammatio cutis racemosa », télangiectasies généralisées ou « asphyxia reticularis multiplex ».
 La dénomination de cutis marmorata, encore d’usage courant pour désigner les livedos du nouveau-né, devrait être abandonnée dans un souci de simplification de la nomenclature dermatologique.
La dénomination de cutis marmorata, encore d’usage courant pour désigner les livedos du nouveau-né, devrait être abandonnée dans un souci de simplification de la nomenclature dermatologique.
Ce terme de cutis marmorata est parfois cependant encore utilisé dans la littérature anglo-saxonne pour désigner les livedos physiologiques.
Le livedo est une manifestation cutanée fréquente, le plus souvent sans aucune signification pathologique.
Ailleurs, il constitue un symptôme qu’il importe de prendre en considération du fait de ses causes variées, parfois responsables d’atteintes systémiques graves.
L’interrogatoire et l’examen clinique sont les éléments-clés du diagnostic étiologique.
Physiopathologie et différents aspects cliniques :
Depuis les travaux de Renaut (1883) et Unna (1896), l’unité vasculaire fonctionnelle cutanée est considérée comme une zone en forme de cône dont la base en forme de cercle ou d’ovale se trouverait à la surface de l’épiderme et dont le sommet correspondrait à une artériole nourricière du derme profond.
Entre les différentes unités vasculaires contiguës existerait un dense réseau anastomotique superficiel et profond.
La partie centrale du cône à la surface du revêtement cutané serait la zone de circulation maximale alors que la périphérie aurait une circulation réduite.
Les mesures de la température cutanée mettent d’ailleurs en évidence une diminution de 0,1 °C du centre à la périphérie des cercles en surface.
Le livedo est la traduction clinique d’une vasodilatation des vaisseaux de surface ; cette vasodilatation touche de façon prédominante tantôt le réseau veineux, d’où la teinte érythrocyanique de certains livedos, ailleurs le versant capillaire, d’où leur teinte érythémateuse.
Il est classique d’opposer les livedos vasomoteurs secondaires à des anomalies de la vasoconstriction sans anomalie pariétale ou endoluminale vasculaire aux autres livedos comportant une atteinte vasculaire anatomique pouvant s’accompagner secondairement d’anomalies vasomotrices de voisinage liées à l’ischémie.
L’atteinte vasculaire anatomique touche principalement les artérioles du derme profond et/ou de l’hypoderme mais aussi les vaisseaux de plus petit calibre du derme superficiel ou moyen, parfois isolément.
Théoriquement, l’atteinte artériolaire touche principalement l’artère nourricière du cône se projetant en surface sur la zone apparemment saine entre les mailles du livedo.
Ainsi, dans le syndrome de Sneddon, les anomalies anatomopathologiques seraient, d’après certains auteurs, principalement observées sur les biopsies profondes prélevées au centre des mailles.
Cette théorie est remise en cause par l’expérience clinique, notamment dans d’autres dermatoses lorsqu’il existe des zones infiltrées ou des zones nécrotiques observées préférentiellement sur les mailles du livedo.
Il est alors préférable de biopsier ces zones infiltrées ou nécrotiques pour visualiser les vaisseaux pathologiques.
C’est le cas des livedos de la périartérite noueuse (PAN) ou des thrombocytémies.
L’aspect du livedo dépend également de son mécanisme physiopathologique ou de l’extension des lésions vasculaires.
En présence d’anomalies vasomotrices ou d’atteinte diffuse artériolaire homogène, le livedo est réticulé avec des mailles fermées et régulières ; à l’opposé, le livedo ramifié ou « racemosa » à branches ouvertes, non fermées, traduit une atteinte artériolaire irrégulière.
Dans la littérature européenne, la distinction entre le livedo réticulé à mailles fines régulières formant des cercles complets et le livedo ramifié à mailles irrégulières formant des ramifications ou des cercles incomplets est généralement précisée.
En revanche, dans la littérature anglo-saxonne, le même terme « livedo reticularis » est employé pour tout livedo pathologique, que ses mailles soient régulières ou non, d’où les difficultés d’analyse sémiologique de certains articles.
Quel que soit le type du livedo, il est principalement visible sur peau blanche et rare sur peau noire.
Diagnostic différentiel :
De nombreuses dermatoses, d’origine non vasculaire, peuvent prendre un aspect réticulé simulant ainsi un livedo.
Celles-ci sont en général érythématosquameuses, purpuriques ou pigmentées alors que le livedo est érythémateux ou cyanotique disparaissant à la vitropression.
La plus fréquente dermatose simulatrice de livedo est l’érythème « a calore », également appelé « pigmentation des chaufferettes ».
Il se développe sur les zones cutanées, chroniquement exposées à une source de rayonnement infrarouge.
Initialement, il s’agit d’un érythème réticulé transitoire, se transformant secondairement en un réseau à larges travées pigmentées ne s’atténuant que très lentement après cessation de l’exposition.
La photosensibilisation induite par la quinidine apparaît 2 à 48 heures après une exposition solaire, se manifestant par un oedème prurigineux avec une éruption réticulée gris-bleu devenant purpurique puis pigmentée, ne s’effaçant pas à la vitropression.
Les lésions sont localisées sur les zones exposées épargnant relativement le visage et les extrémités.
La dose érythémateuse minimale avec les ultraviolets A est abaissée.
D’autres dermatoses peuvent prendre un aspect réticulé telles que la mucinose érythémateuse réticulée de Steigleder, les poïkilodermies héréditaires ou acquises, le lichen plan ou d’autres éruptions lichénoïdes, les lésions lupiques, la mélanose de Riehl, la papillomatose confluente et réticulée de Gougerot et Carteaud, les syphilides, l’incontinentia pigmenti, les eczematide-like purpuras, l’urticaire pigmentaire…
Ces dermatoses sont en règle générale aisément reconnaissables du fait de leur aspect clinique et de leurs caractéristiques anatomopathologiques.
Diagnostic étiologique :
Il est orienté par les caractéristiques cliniques du livedo, la présence d’autres lésions dermatologiques et le contexte clinique.
Sont à prendre en considération l’âge d’apparition du livedo, sa topographie sur les membres ou sur le tronc, voire sur le visage, son caractère déclive ou suspendu, la présence d’une éventuelle infiltration qui doit être soigneusement recherchée par la palpation de toutes les mailles ainsi que son aspect réticulé ou ramifié à mailles fines ou larges.
Les variations du livedo en fonction de la position debout ou allongée du sujet ainsi que de la température sont également importantes à noter en sachant que les livedos permanents sont toujours pathologiques alors que les livedos n’apparaissant qu’en position debout ou dans des conditions thermiques particulières ne sont pas toujours physiologiques.
En effet, de nombreux livedos pathologiques sont accentués par la position déclive et le froid.
A priori, tout livedo d’apparition tardive sans perte de poids importante, de type ramifié, suspendu et/ou infiltré peut être considéré comme pathologique.
La présence d’autres lésions dermatologiques telles qu’une zone nécrotique, une ulcération, des lésions purpuriques, des nodules, des lésions atrophiques ou cicatricielles, des hémorragies multiples en « flammèches » sous-unguéales, un orteil pourpre constitue également un argument de poids en faveur du caractère pathologique du livedo.
Le contexte clinique est également très important à connaître dans cette démarche diagnostique.
Aussi seront soigneusement notés les prises médicamenteuses, tous les antécédents cliniques personnels ou familiaux, l’interrogatoire étant plus particulièrement orienté vers les antécédents cardiovasculaires (hypertension artérielle [HTA], valvulopathie, athérosclérose…), neurologiques, oculaires, gynécoobstétricaux (nombre de fausses couches spontanées), néphrologiques, digestifs… Un examen clinique complet est indispensable.
En fait, plusieurs mécanismes peuvent être présents simultanément ; ainsi, les obstructions artériolaires aiguës peuvent s’accompagner de phénomènes de vasodilatation réflexe de voisinage par ischémie…
A – LIVEDOS VASOMOTEURS :
Lorsque des anomalies de la vasomotricité sont le mécanisme physiopathologique principal du livedo, ce dernier est habituellement réticulé et régulier.
1- Livedo physiologique du nouveau-né :
Appelé également cutis marmorata simple, ce livedo, surtout visible aux extrémités, apparaît lorsque le nouveau-né est exposé au froid et disparaît lors du réchauffement.
Il s’agit d’un phénomène vasculaire physiologique, fréquent et transitoire, ne s’accompagnant jamais de troubles trophiques.
2- Livedo idiopathique dit de stase ou physiologique :
Il s’agit d’une cyanose réticulée des membres ; il est observé préférentiellement chez la femme jeune, favorisé par le froid et l’orthostatisme (livedo déclive).
Ce livedo est parfois permanent, plus étendu, alors volontiers associé à une acrocyanose avec hypersudation et parfois à une anorexie.
Il s’atténue souvent spontanément ou persiste sans signification pathologique.
3- Livedo et amantadine :
Un livedo réticulé des extrémités est observé chez 30 % des parkinsoniens traités par amantadine, résultant d’une vasoconstriction artériolaire secondaire à la libération neuronale de dopamine et de catécholamines induite par ce médicament.
Il n’impose pas l’arrêt du médicament et régresse en 4 semaines après la fin du traitement.
Il n’y a pas de parallélisme entre l’amélioration des signes neurologiques et l’apparition du livedo.
La maladie neurologique n’a d’ailleurs aucune implication dans l’apparition de ce livedo puisqu’il est également observé chez les sujets témoins indemnes de toute affection, prenant de l’amantadine au long cours.
4- Livedo et phéochromocytome :
Il s’agit d’une manifestation exceptionnelle de phéochromocytome.
Le livedo prédomine sur les extrémités, généralement associé à une peau moite avec, lors des crises hypertensives paroxystiques, des sueurs profuses et une pâleur extrême. Le diagnostic est confirmé par le dosage des catécholamines et de leurs métabolites urinaires.
5- Livedo et maladies neurologiques :
Un livedo réticulé, probablement par stase, peut être observé sur un membre dont la mobilité est réduite du fait d’une atteinte neurologique périphérique ou centrale.
Ailleurs, les anomalies de la vasomotricité des vaisseaux cutanés seraient liées à une atteinte neurologique centrale cérébrale ou médullaire avec apparition d’un livedo sur des zones non paralysées.
Ce mécanisme physiopathogénique pourrait expliquer certains livedos constatés au cours de la sclérose en plaques, de poliomyélites, d’encéphalites, de maladie de Parkinson ou d’autres atteintes neurologiques centrales.
6- Livedo et bas débit circulatoire :
L’apparition d’un livedo au cours des chocs cardiogéniques ou hypovolémiques avec un bas débit circulatoire est généralement de mauvais pronostic.
Il en est de même des marbrures observées au cours des états de choc toxi-infectieux.
Quant au livedo des pancréatites, il a été exceptionnellement rapporté au cours de crises de pancréatite avec douleurs, vomissements abondants et collapsus.
L’hypothèse d’une action directe des enzymes pancréatiques sur la paroi des vaisseaux n’a jamais été démontrée.
B – LIVEDOS SECONDAIRES À UNE OBSTRUCTION VASCULAIRE :
Les livedos secondaires à une perturbation circulatoire locale obstructive sont associés à des lésions anatomiques pariétales ou endoluminales des vaisseaux du derme et/ou de l’hypoderme.
Nous avons schématiquement distingué les vasculites, les thromboses et les embolies, ces différents mécanismes pouvant être associés comme dans les embolies de cristaux de cholestérol (embolie, vasculite, thrombose).
1- Vasculites :
Théoriquement, toutes les vasculites peuvent s’associer à la présence d’un livedo, quelle qu’en soit la cause.
Celle-ci ne permet pas de préjuger du type de vaisseaux atteint ; en effet, si elle traduit le plus souvent une atteinte des artérioles de moyen calibre, elle a également été rapportée dans les vasculites des veinules postcapillaires du derme superficiel ou profond.
Au sein du vaste spectre des vasculites, le livedo est surtout observé au cours de la PAN et des cryoglobulinémies.
* Périartérite noueuse :
Le livedo de la PAN est ramifié, suspendu, localisé principalement aux membres inférieurs, parfois aux membres supérieurs.
Habituellement, la palpation soigneuse permet de repérer quelques zones infiltrées qui doivent être biopsiées au bistouri pour mettre en évidence la vasculite nécrosante des artérioles hypodermiques.
Une vasculite des petits vaisseaux peut être associée, exceptionnellement isolée.
D’autres manifestations dermatologiques telles que des nodules en dehors des zones de livedo, des nécroses cutanées, des ulcérations, un purpura, des oedèmes segmentaires, témoignent du polymorphisme de l’expression clinique de cette vasculite.
La prévalence du livedo dans les PAN cutanées isolées est de l’ordre de 56 à 78 % des cas.
Dans leur grande majorité, ces PAN cutanées ont une évolution chronique sans risque vital ; l’association à une neuropathie sensitive très algique ne modifie pas leur pronostic vital.
Ailleurs, elles peuvent représenter le tableau initial d’une PAN systémique qui seule justifie une corticothérapie générale.
La PAN systémique a été récemment démembrée en PAN classique, caractérisée par une atteinte des artères de moyen calibre comportant une nécrose fibrinoïde pariétale et polyangéite microscopique caractérisée par une atteinte des petits vaisseaux (capillaires, veinules ou artérioles) avec fréquemment présence d’anticorps anticytoplasme des polynucléaires neutrophiles de type périnucléaire dirigés contre la myéloperoxydase.
Dans les anciennes séries, le livedo était présent dans seulement moins d’un tiers des cas de PAN systémiques, le purpura étant ici la plus fréquente des lésions dermatologiques.
Il est probable que sa prévalence soit plus élevée dans la PAN classique.
* Cryoglobulinémies :
Une vasculite est principalement observée au cours des cryoglobulinémies de type II et III avec présence d’un livedo respectivement dans 22 et 20 % des cas.
Le livedo est habituellement associé à d’autres manifestations dermatologiques et en particulier à un purpura vasculaire, plus rarement à des ulcérations plus ou moins nécrotiques. Histologiquement, la vasculite prédomine sur les veinules du derme superficiel ou profond.
L’évolution en est volontiers chronique avec possibilité de manifestations articulaires et d’une atteinte viscérale (rénale ou neurologique).
Ces cryoglobulines peuvent être symptomatiques d’affections auto-immunes ou d’infections virales.
Il a été récemment démontré que le virus de l’hépatite C est responsable de plus de la moitié des formes autrefois considérées comme idiopathiques.
Parfois cependant, aucune cause ne peut être mise en évidence.
2- Thromboses des vaisseaux cutanés :
Toute thrombose des vaisseaux cutanés peut se manifester par un livedo, parfois isolé au début, le plus souvent associé à d’autres lésions dermatologiques, souvent purpuriques et nécrotiques.
La biopsie du livedo ne révèle qu’inconstamment la thrombose alors que celle réalisée sur le liseré purpurique d’une zone nécrotique permet habituellement de confirmer le diagnostic de thrombose sans préjuger du diagnostic étiologique.
* Atrophie blanche idiopathique :
L’atrophie blanche idiopathique ou vasculite hyalinisante segmentaire (livedoid vasculitis des Anglo-Saxons) est souvent prise à tort pour une vasculite.
La lésion primitive est purpurique, évoluant vers une nécrose très douloureuse ; la cicatrisation, lente, laisse place à des lésions séquellaires, porcelainées et atrophiques avec une bordure télangiectasique et pigmentée.
Les lésions sont limitées aux membres inférieurs.
Le livedo est inconstant, uniquement localisé dans les zones périulcéreuses ou plus diffus, réticulé ou ramifié, non infiltré.
La biopsie doit porter sur un élément purpurique récent.
Elle permet de visualiser des thrombi hyalins des vaisseaux dermiques sans infiltrat inflammatoire notable.
L’immunofluorescence directe est inconstamment positive avec des dépôts vasculaires de fibrine dans les lésions récentes et d’immunoglobulines (Ig) M, d’IgG et de C3 dans les lésions plus anciennes.
L’étiopathogénie de l’atrophie blanche reste obscure, probablement non univoque.
Ont été invoquées une baisse de l’activité fibrinolytique, une augmentation de l’activité plaquettaire, des anomalies de la cellule endothéliale avec diminution de l’expression de la thrombomoduline ou une anomalie de la coagulation.
Certaines observations ont été intégrées dans le syndrome des antiphospholipides (SAPL), la prévalence des anticorps antiphospholipides dans cette affection demeurant encore inconnue.
Une observation a été décrite en association avec un déficit hétérozygote en protéine C.
Le traitement n’est pas codifié et reste décevant. Ont été essayés avec succès, dans des cas isolés ou de petites séries, l’association héparine, ticlopidine, aspirine ou dipyridamole, la pentoxifylline, un activateur tissulaire du plasminogène recombinant à faible dose en intraveineux (Actilyset), la prostacycline ou ses analogues, la PUVAthérapie ou le danazol à 200 mg/j en traitement prolongé.
Il est tout à fait possible que l’efficacité thérapeutique de tel ou tel médicament dépende du processus pathogénique en cause.
* Coagulation intravasculaire disséminée :
Un livedo d’apparition aiguë peut annoncer l’apparition de lésions nécrotiques au cours des coagulations intravasculaires disséminées, quelle qu’en soit l’origine.
Il est localisé en amont des lésions purpuriques, pouvant remonter sur les jambes, le tronc et les avantbras.
Des tableaux voisins peuvent être induits par des médicaments administrés par voie buccale ou intraveineuse.
Ainsi en est-il de la vasopressine, de la terlipressine ou de l’association diphénhydramine et pyrithyldione….
Une microangiopathie thrombotique peut également être provoquée par la prise de ciclosporine ou de gemcitabine au long cours.
* Déficit en protéines C, S, antithrombine III et anticoagulants :
Toutes les nécroses cutanées plus ou moins extensives peuvent être accompagnées d’un livedo aigu entourant généralement les lésions purpuriques puis nécrotiques.
Ainsi en est-il des nécroses cutanées extensives néonatales en rapport avec un déficit homozygote en protéine C et plus rarement en protéine S, des nécroses cutanées aux antivitamines K associées aux déficits hétérozygotes ou acquis des protéines C ou S, des nécroses induites par l’héparine, souvent associées à une thrombopénie.
Au cours des déficits en protéine C et antithrombine III ont également été décrits des tableaux de livedo chronique plus ou moins étendu, sans autre manifestation dermatologique, associé à des thromboses profondes notamment cérébrales pouvant simuler un syndrome de Sneddon.
* Cryopathies :
Les cryopathies sont à l’origine de nécroses cutanées, de purpura, d’une acrocyanose, d’un syndrome de Raynaud.
Le livedo est présent dans 17 % des cryoglobulinémies de type I, 10 % des maladies des agglutinines froides et occasionnellement signalé au cours des cryofibrinogénémies symptomatiques sporadiques ou familiales.
Alors que les nécroses siègent préférentiellement sur les extrémités (pieds, doigts, oreilles, nez), le livedo prédomine parfois sur les zones découvertes, s’atténuant à la chaleur ; ailleurs, il est généralisé, ramifié.
* Syndrome des antiphospholipides :
La prévalence du livedo dans le SAPL a été estimée de 4 à 55% lorsque le SAPL est primitif et de 9 à 50%lorsqu’il est associé à un lupus systémique.
Sa prévalence n’est pas significativement différente dans l’étude européenne multicentrique comparant 58 malades atteints de SAPL primaire et 56 malades atteints de SAPL associé au lupus.
La fréquente mise en évidence d’anticorps antiphospholipides chez les malades lupiques avec livedo avait été signalée par Hughes dès 1983 et confirmée secondairement.
Le livedo associé au SAPL est habituellement fin, ramifié, suspendu, étendu ou localisé sur plusieurs zones non contiguës, isolé sans autre lésion dermatologique.
Une thrombose des vaisseaux dermiques ou hypodermiques est alors exceptionnellement objectivée.
L’examen anatomopathologique d’un prélèvement cutané entre les mailles ou sur les mailles ne met en effet en évidence le plus souvent qu’une hyperplasie vasculaire ou des lésions d’endartérite oblitérante non spécifique.
Ailleurs, il peut être localisé entourant une zone nécrotique ou aux extrémités, d’évolution plus aiguë, notamment en cas de syndrome catastrophique du SAPL avec présence d’autres lésions dermatologiques (nécrose, gangrène, lésions bulleuses ou érythème palmaire) et objectivation d’une thrombose histologique.
Que le SAPL soit primaire ou associé au lupus, la présence d’un livedo ramifié est statistiquement associée non seulement aux accidents thrombotiques artériels, en particulier aux accidents vasculaires cérébraux, mais aussi aux valvulopathies et à l’HTA.
L’association livedo-accidents vasculaires cérébraux pose le problème des relations du syndrome de Sneddon avec le SAPL.
* Syndromes myéloprolifératifs :
Les thrombocytémies essentielles ou secondaires à un autre syndrome myéloprolifératif (maladie de Vaquez, leucémie myéloïde ou splénomégalie myéloïde) sont parfois à l’origine d’un livedo ramifié douloureux, symétrique et distal, parfois inaugural.
Il s’y associe souvent d’autres manifestations dermatologiques : syndrome de Raynaud, érythrocyanose, érythromélalgie, ulcérations nécrotiques punctiformes sur les mailles du livedo, voire gangrènes distales.
Les biopsies cutanées sur les mailles du livedo objectivent le plus souvent des thromboses vasculaires.
L’association à des thromboses profondes, notamment cérébrales, a pu faire porter à tort le diagnostic de syndrome de Sneddon dont le livedo est différent du fait de sa topographie volontiers tronculaire et de l’absence habituelle d’algies ou de nécrose.
* Calcinoses cutanées :
Les dépôts de calcium dans la peau s’accompagnent souvent de nécroses cutanées souvent extensives précédées ou accompagnées d’un livedo d’évolution aiguë.
Ces dépôts peuvent être secondaires à une panniculite ou à une anomalie du métabolisme phosphocalcique telle qu’une hyperparathyroïdie primitive ou induite par une insuffisance rénale chronique.
Les mécanismes physiopathologiques conduisant au livedo et à la nécrose sont multiples faisant intervenir les dépôts vasculaires de calcium, des thromboses avec possibilité d’anomalies acquises de la coagulation, un spasme vasculaire.
Le diagnostic est généralement évident sur les biopsies cutanées prélevées de préférence sur les zones indurées ou nécrotiques.
Le pronostic est encore sombre avec nécessité de corriger au plus vite un éventuel déséquilibre du bilan phosphocalcique.
* Oxalose :
L’oxalose est le plus souvent liée à une maladie métabolique héréditaire autosomique récessive conduisant à une insuffisance rénale terminale par lithiases oxaliques récidivantes.
Les sels d’oxalate de calcium peuvent être retrouvés dans les parois vasculaires et la peau, se manifestant notamment par une acrocyanose, des gangrènes périphériques et un livedo des membres inférieurs.
La biopsie sur les mailles révèle la présence de cristaux biréfringents en lumière polarisée, siégeant dans le derme et la paroi des vaisseaux souvent thrombosés.
De nombreuses manifestations sytémiques pouvant simuler une vasculite témoignent de la diffusion de ces dépôts.
3- Embolies :
Habituellement, c’est le contexte clinique qui fait évoquer un processus emboligène, notamment en présence d’une athérosclérose.
Plus rarement, ce contexte est méconnu, le livedo pouvant être révélateur d’une cardiopathie emboligène, tel un myxome.
* Embolies de cristaux de cholestérol :
Les embolies multiples de cristaux de cholestérol ont une expression cutanée dans 35 à 45 % des cas. Le livedo en est la manifestation la plus fréquente, présent chez 49 à 90 % des malades avec atteinte cutanée.
Habituellement, il est ramifié, localisé aux membres inférieurs, pouvant s’étendre sur les lombes et sur le bas de l’abdomen, exceptionnellement aux membres supérieurs.
Il peut n’apparaître qu’en position debout.
Il est généralement associé à d’autres manifestations cutanées : nécroses sur les mailles ou à distance, gangrènes, cyanose, ulcérations, purpura, orteils pourpres.
Ce diagnostic est systématiquement évoqué chez un sujet athéromateux, et ce d’autant plus qu’il existe un syndrome algique, des pouls conservés, des manifestations systémiques pouvant simuler une PAN (atteinte rénale, digestive, altération de l’état général) et un facteur déclenchant, tels un geste chirurgical vasculaire, un cathétérisme artériel et/ou la mise en route d’un traitement anticoagulant (héparine, antivitamines K) ou fibrinolytique.
Le diagnostic repose sur la mise en évidence des cristaux de cholestérol au fond d’oeil et surtout dans les artérioles cutanéomusculaires.
Étant donné la localisation des cristaux dans les artérioles de la jonction dermohypodermique, il est nécessaire de faire des biopsies cutanées profondes, de préférence sur les zones infiltrées ou nécrotiques du livedo et de les répéter si elles sont non contributives.
Le risque de nécrose secondaire à ces biopsies est minime en l’absence d’atteinte artérielle majeure des gros troncs jambiers.
Un prélèvement effectué en peau saine n’a pas d’intérêt.
À l’opposé, une biopsie musculaire quadricipitale à l’aveugle serait positive dans 92 à 100 % des cas.
En cas d’atteinte rénale, la rentabilité de la biopsie rénale est également élevée mais non dénuée de risque.
Quelle que soit l’origine du prélèvement, la mise en évidence sur une biopsie d’une vasculite ou d’une thrombose isolée ne doit pas faire rejeter le diagnostic d’emboles de cholestérol du fait d’une association possible.
L’étendue du spectre clinique des embolies de cristaux de cholestérol, allant des formes cutanées isolées aux formes systémiques rapidement mortelles, explique les difficultés de l’analyse prospective ou rétrospective des traitements.
Il n’existe aucune preuve d’efficacité des traitements médicaux.
Aussi, les traitements sont-ils essentiellement symptomatiques.
Les formes cutanées limitées s’améliorent souvent spontanément avec des soins locaux éventuellement associés à une hémodilution, voire à des dérivés des prostacyclines en cas de nécrose distale pour tenter de limiter les amputations.
L’HTA est traitée par des inhibiteurs de l’enzyme de conversion de l’angiotensine.
L’effet bénéfique des corticoïdes dans les formes systémiques a été signalé avec même une corticodépendance dans certaines observations.
Théoriquement, les anticoagulants doivent être arrêtés car ils sont considérés comme des facteurs favorisants.
Or, leur arrêt n’influence l’évolution des emboles de cristaux que dans des observations privilégiées.
Leur maintien lorsqu’ils sont indispensables ne s’accompagne pas systématiquement d’une aggravation du tableau clinique.
La prudence justifie cet arrêt dans tous les cas où l’indication n’en est pas impérative.
Les antiagrégants ne semblent pas avoir les mêmes inconvénients.
La place du traitement chirurgical est encore controversée.
Dans l’hypothèse d’un anévrisme aortique ou poplité, la découverte de cette anomalie à l’occasion d’embolies de cristaux ne modifie pas l’indication chirurgicale habituellement préconisée.
En l’absence d’anévrisme, la source des embolies reste généralement hypothétique, même en cas de visualisation de plaques d’athérosclérose, d’ailleurs souvent multiples, et la chirurgie est souvent plus dangereuse que bénéfique.
* Myxomes :
Les myxomes cardiaques simulent également parfois une vasculite systémique, plus rarement un syndrome de Sneddon.
Ils peuvent se manifester par un livedo, volontiers distal et associé à d’autres manifestations dermatologiques : syndrome de Raynaud, éruption papuleuse parfois serpigineuse, papulonodules des extrémités, lésions purpuriques et nécrotiques, hémorragies en « flammèches » sous-unguéales.
La biopsie cutanée, faite sur les mailles et entre les mailles, objective inconstamment les embolies myxomateuses. Parfois ne sont observées qu’une vasculite ou des thromboses.
Le rôle de la production d’interleukine 6 par les cellules myxomateuses ou de l’exceptionnelle présence d’anticorps antiphospholipides est encore hypothétique.
Toutes les manifestations cliniques régressent après exérèse de la tumeur.
* Dermite livédoïde de Nicolau :
Décrite en 1925 par Nicolau à la suite de l’injection intramusculaire de sels de bismuth, cette dermatose était initialement considérée comme secondaire à une injection intra-artérielle accidentelle d’un produit huileux.
Actuellement, il est admis que toute intramusculaire peut déclencher une telle dermatose, même en cas d’injections périartérielles.
L’injection s’accompagne d’une douleur intense et sévère suivie de l’apparition d’une zone livédoïde avec une évolution nécrotique.
La physiopathologie fait intervenir des phénomènes emboliques et thrombotiques associés à un vasospasme d’origine endothéliale ou neurologique.
Les antiagrégants, anticoagulants et vasodilatateurs n’empêchent pas toujours l’évolution vers une nécrose plus ou moins étendue.
* Tumeurs métastatiques :
Exceptionnellement, des carcinomes métastatiques peuvent se manifester par un livedo de mécanisme embolique avec envahissement des lymphatiques dermiques objectivé sur la biopsie cutanée.
* Maladie des caissons :
Au cours de la maladie des caissons existe souvent un livedo plus ou moins étendu, éphémère, pouvant s’associer à une sensation de fourmillement localisé ou généralisé, n’ayant pas de signification pronostique quant à la possibilité de survenue de manifestations plus graves, notamment cardiopulmonaires, neurosensorielles ou ostéoarticulaires.
Lié à l’apparition de bulles d’azote dans les capillaires cutanés, il disparaît spontanément ou après une douche chaude.
4- Lymphome angiotrope :
Le lymphome angiotrope, anciennement dénommé angioendothéliomatose proliférative maligne systématisée, est une entité anatomoclinique rare touchant préférentiellement l’adulte après 60 ans.
Il s’exprime volontiers au niveau cutané par des nodules angiomateux, des plaques infiltrées, un oedème diffus sclérodermiforme parsemé de télangiectasies arborescentes, plus rarement par un livedo localisé infiltré.
Il s’y associe fréquemment des épisodes neurologiques déficitaires centraux sensitivomoteurs, une fièvre et une asthénie.
Le diagnostic est histologique avec présence de cellules lymphoïdes atypiques B ou T dans les vaisseaux dermiques dilatés.
C – LIVEDOS DE MÉCANISME ENCORE INCERTAIN :
Dans certaines entités, le mécanisme du livedo reste encore incertain, possiblement hétérogène.
Il est toutefois probable que le mécanisme pathogénique prédominant soit des anomalies de la vasomotricité dans le cutis marmorata telangiectatica et une obstruction vasculaire inflammatoire ou non inflammatoire dans le syndrome de Sneddon.
1- Cutis marmorata telangiectatica :
Il se présente dès la naissance comme une érythrocyanose réticulée, persistante après réchauffement contrairement au cutis marmorata simple.
Il est de siège mono- ou dimélique ou généralisé, avec des dilatations vasculaires arborisées ou en « taches ».
Sur ce réseau apparaissent souvent des ulcérations évoluant vers une cicatrice blanche ou atrophique.
L’évolution est variable.
Les formes limitées à la naissance régressent fréquemment précocement dans l’enfance.
Les formes généralisées peuvent persister à l’âge adulte.
Le mécanisme physiopathologique demeure incertain. Une immaturité du système neurovégétatif a été évoquée, peut-être d’origine génétique étant donné l’existence de formes familiales. Dans d’autres formes, l’aspect clinique évoque plus un processus malformatif de type angiome veineux.
Les associations pathologiques seraient fréquentes dans les formes persistantes (50 %) concernant principalement le système cardiovasculaire, le système nerveux central et le système musculosquelettique (Gerritsen MJP, Steijlen PM, Brunner HG, Rieu P. Cutis marmorata telangiectatica congenita: report of 18 cases. Br J Dermatol 2000 ; 142 : 366-369).
Dans le syndrome de Van Lohuizen existent des malformations cardiaques avec notamment persistance du canal artériel.
Le syndrome de Divry-Van Bogaert est caractérisé par une angiomatose cérébrale.
Certains le rapprochent du syndrome de Sneddon, les images d’angiomatose cérébrale pouvant être secondaires aux atteintes vasculaires.
La prédominance masculine et l’existence fréquente de formes familiales différencient cependant ces deux entités.
2- Syndrome de Sneddon :
* Historique – Épidémiologie :
L’association d’un livedo et d’accidents ischémiques cérébraux, décrite initialement par Champion et Rook en 1960, a été authentifié par Sneddon en 1965 à propos de six observations.
Plus de 30 ans après sa description initiale, de nombreuses autres manifestations sont venues enrichir le tableau cutanéoneurologique.
Il s’agit d’une affection rare, diagnostiquée dans 0,26 % des cas d’une série de 3 006 malades avec accidents vasculaires cérébraux.
Inversement, ce diagnostic a été retenu en Allemagne chez 56 % des malades avec livedo ramifié.
En Autriche, l’incidence du syndrome de Sneddon a été estimée à 1 cas par million d’habitants et par an.
Il existe une nette prédominance féminine avec un sex-ratio H/F de 1/3 à 1/6 selon les études.
L’âge moyen au moment du diagnostic est variable, le plus souvent entre 30 et 50 ans alors que les premiers symptômes sont généralement présents depuis de nombreuses années.
La majorité des observations sont considérées comme sporadiques avec cependant une fréquence élevée de manifestations vasculaires ou neurologiques dans la famille.
Les formes familiales sont plus rares avec une agrégation variable, exceptionnellement suggestive d’une transmission autosomique dominante.
* Manifestations cutanées :
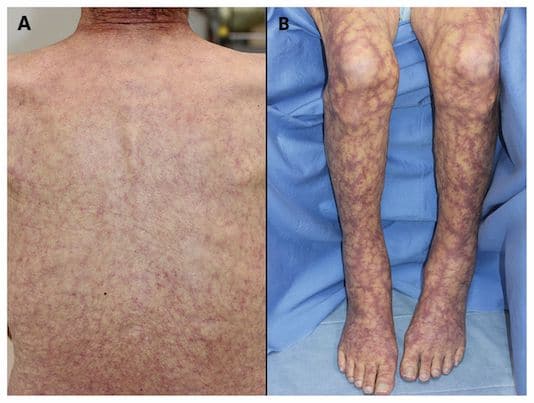
Le livedo du syndrome de Sneddon est un livedo ramifié, généralisé ou étendu à plusieurs territoires non contigus, non déclive.
Il touche très souvent les fesses et le tronc, les membres, parfois le visage, associé parfois à une acrocyanose ou à un syndrome de Raynaud.
À la différence du livedo de la PAN et des autres vasculites, il n’est pas infiltré.
Dans la large majorité des observations, le livedo est la seule manifestation dermatologique.
Ailleurs, il est associé à d’autres lésions : nécrose limitée, ulcération, atrophie blanche, hémorragies sous-unguéales, thrombophlébites superficielles, engelures, etc.
Il précède habituellement de plusieurs années, voire de décennies, l’atteinte neurologique ; plus rarement, il apparaît concomitamment ou plus tardivement. L’aspect anatomopathologique de ce livedo serait pour Zelger et al un élément diagnostique notable.
En effet, 12 fois sur 15, une biopsie de grande taille (2 cm), réalisée au centre des mailles, leur a permis de visualiser, après réalisation de nombreuses coupes sériées, des altérations vasculaires touchant électivement les artérioles de la jonction dermohypodermique qu’ils ont classées en quatre phases histologiques :
– la phase I ou phase initiale correspondrait à une endothélite avec détachement des cellules endothéliales et accolement de cellules mononucléées à leur versant luminal ;
– dans la phase II, l’artériole serait obstruée par des cellules inflammatoires mononucléées ;
– la phase III serait une phase intermédiaire de prolifération cellulaire sous-endothéliale ;
– la phase IV correspondrait à une phase tardive de hyalinisation et de fibroatrophie de la paroi du vaisseau lésé.
A priori, il est difficile de retenir ces différents aspects anatomopathologiques comme un critère diagnostique pour les raisons suivantes.
Dans la littérature, les phases initiales (I, II) n’ont jamais été retrouvées ; quant aux phases III et IV, elles sont certes plus fréquemment observées, présentes chez 21 % des malades dans notre expérience, mais encore moins spécifiques, pouvant être observées après toute obstruction vasculaire, signalées notamment dans diverses atteintes cutanées, rénales et cérébrales du SAPL.
* Manifestations neurologiques :
L’atteinte neurologique est définie par la présence d’accidents ischémiques constitués ou transitoires.
Lorsqu’ils sont constitués, ces accidents vasculaires ont généralement un meilleur pronostic immédiat que les autres accidents vasculaires du sujet jeune du fait de l’atteinte préférentielle des artérioles de moyen calibre.
Lorsqu’ils sont transitoires, le diagnostic différentiel avec certaines migraines ophtalmiques peut être très difficile, d’autant plus que des migraines peuvent parfois être associées et que les accidents vasculaires cérébraux ont fréquemment une localisation occipitale.
La répétition des accidents, parfois infracliniques, peut conduire à long terme à des déficits notables ou à une détérioration intellectuelle, voire à une démence vasculaire. Une épilepsie secondaire est possible comme des accidents hémorragiques, notamment en cas d’anticoagulation.
Une chorée est notée dans quelques observations.
Les techniques modernes d’imagerie, tomodensitométrie et résonance magnétique, visualisent sans peine les infarctus cérébraux de grande et moyenne tailles.
La résonance magnétique révèle également des lésions anoxiques de petite taille (hypersignaux intenses) et des lésions ischémiques hypoxiques (plages confluentes d’hypersignal) prédominant dans les régions cérébrales postérieures avec atteinte du cervelet et des noyaux gris centraux dans les formes évoluées.
L’artériographie cérébrale ou l’imagerie vasculaire non invasive est normale dans près de la moitié des cas ; ailleurs, elles montrent des sténoses ou des obstructions des artères intracérébrales de moyen calibre avec possibilité de développement d’un réseau capillaire en « volutes de fumée ».
Des microemboles ont été dépistés par monitoring de l’artère cérébrale moyenne avec un doppler transcrânien. L’examen du liquide céphalorachidien est sans particularité.
L’électromyogramme systématique des membres inférieurs peut objectiver une neuropathie axonale périphérique généralement totalement asymptomatique.
Les rares études histologiques cérébrales retrouvent une vasculopathie oblitérante non inflammatoire et des thromboses.
Dans un cas a été notée une infiltration granulomateuse sans tropisme vasculaire.
* Autres manifestations :
Une HTA est présente dans plus de 60 % des cas, habituellement modérée, sans relation avec la précocité ni la gravité de l’atteinte neurologique.
Le mécanisme de cette hypertension est le plus souvent inconnu.
Une cause rénovasculaire a été toutefois mise en évidence dans des observations privilégiées avec anticorps antiphospholipides.
La prévalence des valvulopathies semble élevée, mise en évidence dans près de la moitié des cas des séries avec échographie systématique.
Celles-ci se traduisent par un épaississement valvulaire, parfois sans retentissement fonctionnel alors uniquement dépisté en échographie.
Leur mécanisme est inconnu, pouvant résulter du même processus pathologique que l’atteinte endothéliale artériolaire.
Dans certaines observations, une origine infectieuse est évoquée du fait de la précession par un rhumatisme articulaire aigu.
Les autres manifestations témoignent de la diffusion de l’atteinte vasculaire : artériopathie des membres, de l’oeil, du coeur, des reins.
Les manifestations thrombotiques veineuses sont moins fréquentes, signalées cependant dans plus de 15 % des cas.
* Manifestations biologiques :
Le bilan biologique standard est habituellement normal en dehors d’une inconstante augmentation de la vitesse de
sédimentation lors des épisodes neurologiques et d’une possible thrombopénie. L’étude de l’hémostase standard et de la fibrinolyse est habituellement normale.
Dans des cas isolés ont été rapportés une hyperréactivité ou une hyperagrégabilité plaquettaire, une augmentation de la b-thromboglobuline témoignant d’une activation plaquettaire, des anomalies du rapport activateur tissulaire du plasminogène-inhibiteur de l’activateur tissulaire du plasminogène, un déficit en antithrombine III, ou une résistance à la protéine C activée.
Du fait de la rareté des observations, il est vraisemblable que l’association entre le syndrome de Sneddon et ces anomalies soit fortuite.
La recherche d’anticorps antinucléaires est occasionnellement positive, généralement à des taux bas.
La présence d’anticorps antiacide désoxyribonucléique (ADN) pose le problème des limites nosologiques du syndrome de Sneddon avec le lupus.
Il n’y a généralement pas de facteur rhumatoïde, d’anticorps anti-SSA, anti-SSB ou antiribonucléoprotéines (RNP).
Dans quelques observations a été notée une cryoglobulinémie.
La prévalence des anticorps antiphospholipides varie selon les séries de 0 à 85% des cas ; elle est de 41 % dans notre expérience.
Du fait de la grande variation de fréquence des anticorps anti-b2 glycoprotéine 1 suivant les techniques utilisées, la signification de la présence isolée de ces anticorps reste encore incertaine.
Quant aux anticorps anticellules endothéliales, retrouvés chez 35 % des malades, leur signification reste également à déterminer.
La comparaison des malades avec ou sans anticorps antiphospholipides met en évidence une plus grande fréquence d’épilepsie et de thrombopénie chez les malades avec anticorps antiphospholipides.
Les malades sans anticorps antiphospholipides ont fréquemment un livedo à mailles plus larges et mieux visible que ceux avec anticorps antiphospholipides.
* Traitement :
Étant donné la rareté des grandes séries de syndrome de Sneddon, il n’existe pas d’étude contrôlée permettant de conclure à l’efficacité ou à l’inefficacité des divers traitements.
De plus, le délai parfois de plusieurs années entre deux accidents ischémiques cérébraux consécutifs rend difficile l’appréciation des résultats thérapeutiques.
L’utilisation de corticoïdes ou d’immunosuppresseurs sans anticoagulation n’a pas empêché la survenue de nouvelles atteintes neurologiques ; ces traitements auraient même un effet délétère dans quelques cas.
Les antiagrégants ou les anticoagulants au long cours pourraient avoir un effet bénéfique.
Dans le SAPL, une forte anticoagulation aurait un meilleur effet préventif des récidives thrombotiques que l’aspirine avec cependant des effets secondaires plus importants.
Aussi paraît-il licite de traiter les syndromes de Sneddon avec anticorps antiphospholipides par les anticoagulants.
Quant aux syndromes de Sneddon sans anticorps antiphospholipides, un traitement antiagrégant peut être proposé initialement ; le traitement anticoagulant n’étant préconisé que secondairement en cas d’apparition d’un événement clinique neurologique ou de détérioration de l’aspect neuro-imagerique.
Tous ces traitements n’ont aucun effet sur le livedo qui peut apparaître sous anticoagulant.
Dans tous les cas, il est logique de tenter d’éradiquer les facteurs de risque vasculaires associés tels qu’une HTA, un tabagisme ou la prise d’oestroprogestatifs.
Conclusion :
Il est impossible de proposer un arbre décisionnel de conduite devant un livedo car le bilan paraclinique dépend essentiellement des caractéristiques du livedo et du contexte clinique.
Un livedo physiologique ne justifie d’aucun examen.







")

")

{kind=link}