



















Diagnostic clinique :
1- Diagnostic prénatal :
Le risque étant plus élevé lorsque l’X surnuméraire est d’origine maternelle, le caryotype réalisé sur les cellules du liquide amniotique est demandé en cas d’âge maternel avancé et peut révéler le syndrome de Klinefelter.
2- Nouveau-né :
 Le syndrome doit être recherché de principe devant certaines anomalies des organes génitaux externes :
Le syndrome doit être recherché de principe devant certaines anomalies des organes génitaux externes :
micropénis (taille < 3 cm), cryptorchidie bilatérale (nouveau-né à terme), hypospadias dans le cadre d’un pseudo-hermaphrodisme masculin.
Il faut signaler un fait méconnu : la fréquence du poids et du périmètre crânien de naissance inférieurs à la moyenne.
3- Enfance :
Les enfants peuvent être normaux : organes génitaux externes, aspect général, performances scolaires.
Dans d’autres cas, les signes suivants permettent d’orienter le diagnostic : 1. micropénis, volume testiculaire inférieur à la normale ; 2. grande taille, minceur, longues jambes (aspect macroskèle) avec diminution du rapport segment supérieur/segment inférieur ; 3. retard psychomoteur, difficultés scolaires, troubles du caractère, surtout s’ils sont associés aux signes précédents.
Rarement une dysmorphie faciale existe : visage carré, rétrognathie, hypertélorisme, signes qui peuvent être plus évidents chez l’adolescent et l’adulte.
4- Adolescence et puberté :
Il n’existe pas de retard pubertaire, sauf en cas de micropénis ou d’ambiguïté sexuelle découverts à la naissance.
Le déroulement de la puberté est variable ; la séquence la plus fréquente est caractérisée par l’augmentation du volume testiculaire, l’apparition de la pilosité pubienne vers 12 à 13 ans, suivies de la croissance de la verge.
Souvent, les testicules atteignent 3 à 3,5 cm de longueur.
Puis ils subissent un arrêt de développement, suivi d’une régression de leur volume vers 15 à 16 ans.
Ce n’est que vers 18 à 20 ans que leur dureté et leur insensibilité sont très caractéristiques.
Mais tous les intermédiaires peuvent être observés : arrêt plus précoce de l’évolution ou au contraire, régression plus tardive de l’involution des tubules séminifères.
La gynécomastie du syndrome de Klinefelter est un signe fréquent, retrouvé dans 60 à 80 % des cas.
Contrairement à la gynécomastie physiologique, qui apparaît vers 14 ans, en milieu de puberté et s’accompagne de testicules ayant atteint plus de 4 cm de longueur, celle du Klinefelter est souvent plus tardive, vers 16 à 17 ans, alors que les testicules sont petits.
Le risque de cancer du sein est plus fréquent que chez les hommes normaux.
5- Adulte :
Lorsque la virilisation d’un adolescent a été normale et si la régression du volume testiculaire est incomplète, le diagnostic peut être porté lors d’une consultation pour stérilité ou impuissance.
Certains hommes atteints de ce syndrome ont des testicules de 3,5 cm de long, plus ou moins durs et sensibles : il s’agit souvent de mosaïque 46,XY/47,XXY. C’est aussi en cas de mosaïque que la fertilité peut être préservée.
On pourrait faire un parallèle avec le syndrome de Turner : dans les deux syndromes la stérilité est de règle, avec de rares exceptions.
L’installation d’une obésité est possible à l’âge adulte, non ou insuffisamment corrigée par la testostérone.
Croissance et anomalies squelettiques :
1- Croissance :
Elle est variable ; elle n’est pas aussi bien connue et décrite que dans le syndrome de Turner.
Cependant, ces garçons ont généralement une courbe de croissance au-dessus de la moyenne, mais on ne peut donner de fréquence exacte ; c’est la longueur excessive des jambes qui est la cause de cette grande taille ; elle ne dépend pas de la sécrétion de la testostérone puisqu’elle est observée avant la puberté, mais elle s’exagère à l’adolescence.
À l’inverse, certains enfants ont une taille évoluant au-dessous de la moyenne.
On retrouve ces variations dans l’évaluation de la taille finale : selon les séries, la moyenne varie de 1,75 m à 1,80 m avec des extrêmes de 1,63 m à 1,93 m.
Il faut remarquer que les sujets ayant plus d’un seul X surnuméraire ont fréquemment une taille inférieure à la normale.
2- Anomalies squelettiques :
La plus connue est l’apparition d’une scoliose à l’adolescence.
Les clichés osseux révèlent parfois des hémi-vertèbres, des blocs vertébraux, des pouces triphalangiens.
Les anomalies dentaires sont peu connues mais intéressent les généticiens puisque la croissance des dents est influencée par le dimorphisme sexuel : l’épaisseur de l’émail et de la dentine est inférieure à celle des hommes normaux mais supérieure à celle des femmes.
3- Autres anomalies :
La prédisposition aux tumeurs et aux cancers est reconnue : tératomes médiastinaux responsables de signes de précocité pubertaire avec sécrétion exagérée d’hormone chorionique gonadotrophique (HCG), lymphomes, leucémies, adénome du foie.
La fréquence du diabète sucré est plus élevée que dans la population générale.
La bronchite chronique, les ulcères variqueux, l’ostéoporose sont décrits chez l’adulte.
Quotient intellectuel et comportement :
De grandes variations existent, mais le quotient intellectuel moyen (QI) se situe autour de 90-100 avec des scores parfois élevés (« intelligence supérieure ») ou plus faibles particulièrement si le nombre de chromosomes X augmente.
Le QI est généralement supérieur aux performances verbales (troubles du langage).
Les troubles du comportement sont très variables : calme, apathie, manque d’initiative contrastent avec des crises d’agressivité pouvant confiner, rarement, à de véritables comportements psychiatriques.
Certains sujets, spécialement ceux qui ont plus de 2 chromosomes X, ont été dépistés dans les hôpitaux psychiatriques.
Ce tableau psychoaffectif variable invite donc à rechercher la cause des différences retrouvées et à approfondir l’évolution des performances de l’enfance jusqu’à l’âge adulte, puisque les séries pédiatriques manquent de repères pour apprécier l’évolution chez l’adulte. Mais, il a été démontré que la prise en charge précoce de ces enfants avec des moyens pédagogiques et un soutien psychologique adaptés pouvait considérablement améliorer le retard mental et les troubles du comportement : on retrouverait ainsi dans ce syndrome la même évolution concernant la prétendue faiblesse intellectuelle chez les filles turnériennes.
Diagnostic hormonal :
Avant la puberté, les concentrations de base de FSH et de LH sont normales, mais leur réponse à l’injection de Gn-RH (ou LH-RH) peut être supérieure à la normale.
Au début et au cours de la puberté, les valeurs basales de FSH sont anormalement élevées, de même que la réponse des gonadotrophines à la stimulation par la Gn-RH : cela traduit l’insuffisance de sécrétion progressive de la testostérone.
Cependant, il existe des variations dans l’apparition de cette séquence pathologique hormonale : plus les testicules sont dysgénétiques, plus les altérations du rétrocontrôle entre testicules et hypophyse seront précoces.
À l’inverse, la sécrétion de testostérone peut être longtemps préservée chez un adulte porteur d’une mosaïque 46,XY/47,XXY.
La réponse de la testostérone plasmatique après injection d’HCG est normale avant la puberté, inférieure à la normale au cours ou au décours de la puberté.
On pense que la gynécomastie est due à l’insuffisance de sécrétion de la testostérone associée à une élévation des protéines porteuses SHBG (sex hormone binding globulin), ce qui entraîne une baisse de la concentration de testostérone libre ; l’autre mécanisme est une augmentation du rapport oestradiol/testostérone.
Des anomalies de la fonction thyroïdienne ont été signalées, la montée de la TSH étant parfois insuffisante après stimulation par la TRH (thyrotropin releasing hormone).
Environ 10 % des adultes ont des taux élevés d’anticorps anti-thyroglobuline.
Cependant, l’insuffisance thyroïdienne cliniquement décelable n’est pas la règle.
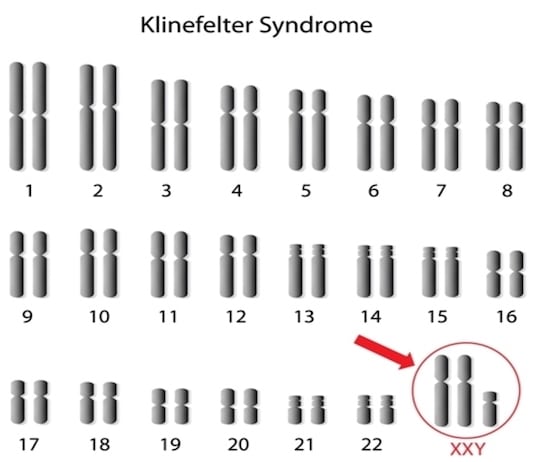
Diagnostic cytogénétique :
Le frottis buccal n’est plus utilisé à titre diagnostique : la chromatine sexuelle est positive et le nombre de corpuscules de Barr observés varie selon le nombre de chromosomes X (1 corpuscule pour 47,XXY, 2 corpuscules pour 48,XXXY).
C’est le caryotype qui permet d’établir le diagnostic. la formule chromosomique 47,XXY est retrouvée dans plus de 90 % des cas.
Comme dans le syndrome de Turner, des mosaïques sont rapportées mais, faute d’études systématiques à partir de différents tissus, on ne peut évaluer leur fréquence.
L’origine de l’aberration chromosomique vient de la non-disjonction des chromosomes sexuels durant la première ou la seconde division méiotiques, soit d’origine paternelle, soit d’origine maternelle.
Plus rarement il s’agit d’une non-disjonction mitotique après la fertilisation.
L’âge maternel avancé favorise le syndrome de Klinefelter, mais pas autant que dans la trisomie 21.


")




")



{kind=link}