



















Définition :
Les infiltrats lymphocytaires cutanés bénins, ou hyperplasies lymphoïdes bénignes, ou pseudolymphomes cutanés, sont un groupe très hétérogène d’affections simulant cliniquement et/ou histologiquement un lymphome cutané, mais dont l’évolution est bénigne.
Classification :
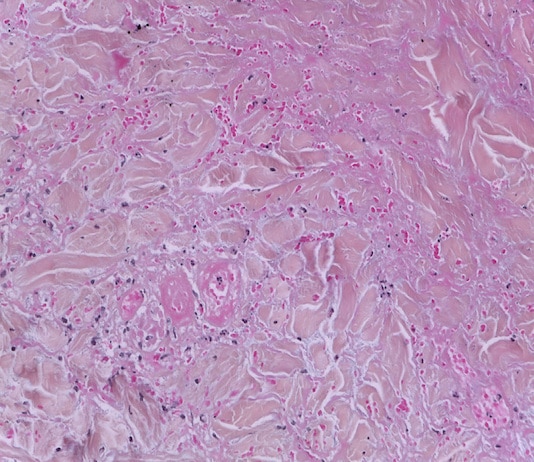 La classification des pseudolymphomes cutanés est fondée sur le phénotype prédominant des lymphocytes de l’infiltrat, la topographie et l’architecture de l’infiltrat, et l’étiologie.
La classification des pseudolymphomes cutanés est fondée sur le phénotype prédominant des lymphocytes de l’infiltrat, la topographie et l’architecture de l’infiltrat, et l’étiologie.
Certaines étiologies peuvent réaliser plusieurs types de tableaux clinique, histologique et immunophénotypique.
Cette classification a un intérêt pratique pour la recherche des différentes étiologies possibles devant un tableau clinique, histologique et immunophénotypique précis.
Les pseudolymphomes cutanés T simulent le plus souvent un lymphome épidermotrope, mais peuvent plus rarement réaliser un infiltrat dermique non épidermotrope, nodulaire.
Dans de nombreux cas, une étiologie est identifiée.
Les pseudolymphomes cutanés B réalisent des infiltrats lymphocytaires denses, nodulaires, bien limités, situés dans le derme superficiel et profond, avec le plus souvent des aspects de centres germinatifs.
Les immunomarquages peuvent révéler la présence de follicules en mettant en évidence le phénotype B de la majorité des lymphocytes centraux, entourés de lymphocytes périfolliculaires de phénotype T.
Les cas sans étiologie retrouvée sont fréquents.
Pseudolymphomes cutanés T :
A – PSEUDOLYMPHOMES CUTANÉS T AVEC INFILTRAT EN BANDE :
1- Pseudolymphomes médicamenteux :
Les pseudolymphomes peuvent être induits par de nombreux médicaments.
Les plus fréquemment en cause sont : anticonvulsivants (barbituriques, carbamazépine, phénytoïne), allopurinol, inhibiteurs de l’enzyme de conversion, bêtabloquants, inhibiteurs calciques, minocycline, méthotrexate, antihistaminiques, ciclosporine, phénothiazine, fluoxétine, hypolipémiants, certains diurétiques.
La plupart de ces médicaments peuvent induire deux types de tableaux bien différents.
Le terme pseudolymphome médicamenteux doit être réservé aux cas de lésions cutanées à type de papules, nodules, tumeurs ou plaques infiltrées, localisées ou disséminées, sans signes généraux.
Les lésions surviennent après 1 à 12 mois de traitement.
L’examen histologique montre un infiltrat lymphocytaire dense, en bande sous-épidermique, épidermotrope, simulant un mycosis fongoïde.
L’absence d’atypies cellulaires, une spongiose épidermique, des nécroses kératinocytaires et des thèques intraépidermiques riches en histiocytes et en polynucléaires éosinophiles sont des arguments en faveur d’un pseudolymphome médicamenteux.
Les immunomarquages montrent que l’infiltrat lymphocytaire est polymorphe, constitué d’un mélange de lymphocytes CD4 et CD8.
Il n’y a le plus souvent pas de clone dominant en biologie moléculaire, mais quelques cas avec présence d’un clone T dans la peau ont été rapportés.
Les lésions cutanées disparaissent en quelques semaines à l’arrêt du médicament inducteur.
Le tableau de pseudolymphome médicamenteux au sens strict doit être distingué des toxidermies à type de syndrome d’hypersensibilité.
Le syndrome d’hypersensibilité survient le plus souvent dans les 2 à 8 premières semaines de traitement, mais le délai peut être de plusieurs années.
Il est plus fréquent chez les patients de peau noire.
Le tableau clinique est caractérisé par la survenue rapide de lésions érythémateuses oedémateuses, souvent prurigineuses ou d’une véritable érythrodermie.
Il peut y avoir une atteinte muqueuse.
Il existe une altération de l’état général avec fièvre, adénopathies, et des atteintes viscérales : hépatosplénomégalie, lésions cardiaques et/ou pulmonaires.
Les anomalies biologiques sont fréquentes : syndrome inflammatoire, hyperleucocytose, lymphocytose avec lymphocytes atypiques, hyperéosinophilie, élévation des lacticodéshydrogénases (LDH), cytolyse hépatique, insuffisance rénale.
L’examen anatomopathologique montre un infiltrat lymphocytaire périvasculaire peu spécifique, un infiltrat polymorphe avec nécroses kératinocytaires évocateur de toxidermie, ou un infiltrat dense épidermotrope simulant un lymphome.
La biologie moléculaire peut mettre en évidence un clone T dans le sang.
L’évolution peut se faire vers le décès du patient malgré une corticothérapie systémique.
2- Dermite de contact lymphomatoïde :
La dermite de contact lymphomatoïde est un eczéma de contact chronique du fait de la persistance de l’allergène, qui simule cliniquement et histologiquement un lymphome épidermotrope.
Les lésions sont des plaques érythématosquameuses parfois eczématiformes évoluant progressivement vers une érythrodermie.
Le prurit est important.
Il peut y avoir des adénopathies.
L’examen histologique montre au début un aspect d’eczéma avec spongiose, mais certains cas réalisent un infiltrat sous-épidermique épidermotrope.
La biologie moléculaire montre le plus souvent l’absence de réarrangement clonal.
L’identification de l’allergène en cause doit être faite grâce à des tests épicutanés orientés par l’interrogatoire. Leur réalisation est souvent difficile en raison du caractère chronique des lésions cutanées.
Parmi les allergènes en cause, l’éthylènediamine a souvent été incriminée.
La corticothérapie locale améliore les lésions mais n’est véritablement efficace qu’après éviction de l’allergène.
3- Lésions secondaires à des piqûres d’insectes et nodules scabieux :
Les piqûres d’insectes ou d’arthropodes peuvent induire des plaques érythémateuses infiltrées ou non, prurigineuses, persistant pendant quelques mois.
Les nodules scabieux sont des papules et des nodules très prurigineux caractérisés par leur localisation aux régions axillaires et génitales, au pli des coudes et à l’ombilic.
Chez le nourrisson, la localisation aux paumes et plantes est évocatrice.
Ces lésions peuvent persister pendant plusieurs semaines ou mois après le traitement antiscabieux.
La cause de cette persistance n’est pas complètement élucidée ; il s’agit vraisemblablement d’une réaction d’hypersensibilité retardée à l’un des constituants du parasite.
Le parasite lui-même n’a été que rarement identifié dans les nodules.
L’examen histologique montre une hyperacanthose épidermique et un infiltrat du derme superficiel et profond constitué de petits lymphocytes, d’histiocytes, de plasmocytes et de polynucléaires éosinophiles souvent associés à quelques cellules de grande taille.
4- Pseudolymphome actinique :
Le pseudolymphome actinique survient habituellement chez l’homme de plus de 50 ans.
Il réalise des placards eczématiformes et des papules rouge violacé confluant en plaques.
Ces lésions infiltrées très prurigineuses débutent sur les zones photoexposées puis s’étendent aux zones couvertes et peuvent ensuite évoluer vers une érythrodermie avec hyperkératose palmoplantaire, alopécie et adénopathies.
Au début, l’examen anatomopathologique peut être non spécifique, compatible avec le diagnostic d’eczéma chronique.
À un stade plus évolué, l’examen anatomopathologique montre une hyperplasie épidermique et un infiltrat lymphocytaire sousépidermique dense, avec exocytose, constitué de lymphocytes atypiques au noyau cérébriforme.
Les immunomarquages montrent que la majorité des lymphocytes de l’infiltrat a un phénotype CD3+ CD8+, ce qui les différencie des lymphocytes du mycosis fongoïde qui ont un phénotype CD3+ CD4+.
Les phototests montrent un effondrement de la dose érythémateuse minimale et une photosensibilité sévère pour un spectre de longueurs d’onde souvent très étendu.
L’évolution se fait vers une aggravation lente avec majoration progressive de la photosensibilité, qui survient pour des expositions de plus en plus minimes, confinant les malades à l’obscurité.
Le pseudolymphome actinique est lié à une sensibilisation rémanente à certains allergènes : plantes compositas, constituants du caoutchouc, bichromate de potassium, parfums, salicylanilides.
Le traitement repose sur l’éviction des allergènes identifiés et la protection solaire rigoureuse par écrans hypoallergéniques.
L’association d’une photothérapie, de corticoïdes et d’azathioprine ou de ciclosporine peut diminuer la sensibilité solaire et aider les patients à mener une vie normale.
5- Papulose lymphomatoïde (type B) :
La papulose lymphomatoïde a été décrite par Macaulay.
Elle réalise des lésions cutanées papulonodulaires érythémateuses régressant spontanément en 3 à 8 semaines.
Certaines lésions évoluent vers une croûte noirâtre évocatrice et guérissent en laissant une cicatrice déprimée parfois pigmentée. D’autres lésions peuvent avoir un aspect folliculaire.
Il peut exister des lésions muqueuses sous forme de papules ou de nodules ulcérés douloureux.
Les lésions peuvent être limitées à quelques éléments sur le tronc ou les membres ou profuses, disséminées sur tout le corps.
Le reste de l’examen clinique est normal.
L’examen anatomopathologique montre un infiltrat dense et épidermotrope.
On distingue plusieurs types de papulose lymphomatoïde.
Dans le type lymphocytaire (type B), il existe un infiltrat dense en bande sous-épidermique, constitué d’une majorité de lymphocytes T à noyau cérébriforme et de quelques cellules de grande taille.
L’aspect histologique de la papulose lymphomatoïde de type histiocytaire (type A) est détaillé dans le chapitre des pseudolymphomes cutanés T avec infiltrat nodulaire.
Ces deux aspects histologiques peuvent coexister chez certains patients. Dans les deux types, il existe des modifications épidermiques : hyperacanthose, spongiose, parakératose, parfois nécrose ou ulcération.
Sur les immunomarquages, les lymphocytes T ont un phénotype CD3+ CD4+, et les cellules de grande taille expriment l’antigène d’activation CD30.
Les lymphocytes de la papulose lymphomatoïde expriment fortement les marqueurs de cytotoxicité (perforine, granzyme, TiA1).
La biologie moléculaire met en évidence un clone T dans les lésions cutanées.
L’évolution se fait sous forme de poussées spontanément régressives pendant quelques mois ou années.
Dans 5 à 15% des cas, la papulose lymphomatoïde peut être précédée, accompagnée ou suivie d’un lymphome (mycosis fongoïde, maladie de Hodgkin ou lymphome T à grandes cellules CD30+).
Dans ces cas, le même clone T a été mis en évidence dans les différents types de lésions.
Cette possibilité d’association justifie une surveillance prolongée des patients.
L’abstention thérapeutique avec surveillance régulière est l’attitude la plus fréquente.
Si cette option est difficile en raison de lésions profuses et/ou subintrantes, on peut envisager un traitement par applications locales de chlorméthine, PUVAthérapie, interféron alpha ou méthotrexate.
L’efficacité peut être difficile à apprécier en raison du caractère spontanément résolutif des lésions.
6- Pseudolymphome associé à une infection par le virus de l’immunodéficience humaine (VIH) :
Ce syndrome survenant chez des malades infectés par le VIH a été rapporté pour la première fois en 1989.
Il s’agit cliniquement d’une érythrodermie avec polyadénopathies et présence de cellules de Sézary dans le sang.
L’examen anatomopathologique montre un infiltrat épidermotrope en bande sous-épidermique constitué de lymphocytes à noyau cérébriforme.
Les caractéristiques particulières de ce tableau sont une photosensibilité, une évolution rapidement pigmentogène et un phénotype CD8.
Il n’y a pas de clone T dominant dans la peau et le sang. Les lymphocytes de l’infiltrat cutané sont oligoclonaux et ont une activité cytotoxique spécifique de certains antigènes du VIH, restreinte par les antigènes de classe I du complexe majeur d’histocompatibilité.
Les combinaisons d’antirétroviraux incluant un inhibiteur de protéase induisent des rémissions complètes et durables.
B – PSEUDOLYMPHOMES CUTANÉS T AVEC INFILTRAT NODULAIRE :
1- Pseudolymphomes médicamenteux :
Certains pseudolymphomes médicamenteux se présentent sous forme de nodules ou de tumeurs érythémateux, uniques ou multiples.
L’examen histologique montre un infiltrat lymphocytaire dense, nodulaire avec ou sans épidermotropisme, simulant un lymphome T pléomorphe à cellules petites et moyennes.
L’absence d’atypies cellulaires, les immunomarquages montrant un infiltrat polymorphe, constitué d’un mélange de lymphocytes CD4 et CD8, et l’absence de clone en biologie moléculaire, sont des arguments en faveur du diagnostic de pseudolymphome.
Les lésions cutanées disparaissent en quelques semaines à l’arrêt du médicament inducteur.
2- Lésions secondaires à des piqûres d’insectes et nodules scabieux :
Les piqûres d’insectes et les nodules scabieux peuvent réaliser des papules ou des nodules érythémateux, prurigineux, simulant un prurigo.
La localisation en régions axillaire, génitale, ombilicale et aux coudes est évocatrice de nodules scabieux.
Les lésions peuvent persister pendant plusieurs semaines ou mois malgré un traitement antiscabieux correctement réalisé et/ou une corticothérapie locale.
Le parasite n’a été que rarement identifié dans les nodules, mais il existe probablement une réaction d’hypersensibilité retardée à l’un de ses constituants.
L’examen histologique montre un infiltrat nodulaire plus ou moins épidermotrope constitué de petits lymphocytes souvent associés à quelques grandes cellules.
3- Papulose lymphomatoïde (type A) :
Le tableau clinique et évolutif de la papulose lymphomatoïde, décrit plus haut dans le chapitre concernant les pseudolymphomes cutanés T avec infiltrat en bande, est identique quel que soit l’aspect histologique.
Les deux types histologiques peuvent être associés chez un même malade.
L’aspect histologique de la papulose lymphomatoïde de type histiocytaire (type A) est caractérisé par un infiltrat plongeant plus profondément dans le derme.
Cet infiltrat est polymorphe, constitué de cellules atypiques de grande taille exprimant l’antigène CD30, mélangées à des polynucléaires neutrophiles et éosinophiles, des histiocytes et des lymphocytes.
Il s’associe à des modifications épidermiques à type d’hyperacanthose, de spongiose, de parakératose, parfois de nécrose ou d’ulcération.
La papulose lymphomatoïde à grandes cellules, ou papulose lymphomatoïde de type C, est caractérisée par un infiltrat constitué quasi exclusivement de nappes de grandes cellules CD30+.
Cet aspect anatomopathologique est identique à l’aspect réalisé par les lymphomes T à grandes cellules CD30+, de type anaplasique ou pléomorphe à grandes cellules. Dans ces cas, c’est la confrontation des données cliniques, anatomopathologiques et évolutives qui permet de porter un diagnostic.
Des lésions papuleuses ou papulonodulaires, parfois nécrotiques, disséminées, d’évolution toujours régressive, font porter le diagnostic de papulose lymphomatoïde.
À l’inverse, une lésion unique de grande taille, tumorale, parfois ulcérée, fait porter le diagnostic de lymphome à grandes cellules.
Dans ce cas, un bilan d’extension à la recherche de localisations extracutanées doit être réalisé, même si la lésion régresse partiellement ou totalement.
Des aspects de papulose lymphomatoïde de type C ont été rapportés après piqûre d’insecte ou dans des nodules scabieux.
4- Infiltration lymphocytaire de Jessner et Kanoff :
L’infiltration lymphocytaire de Jessner et Kanoff se présente sous forme de plaques érythémateuses arciformes du visage, du haut du dos ou de la face antérieure du thorax.
L’évolution se fait par poussées successives avec régression sans cicatrice.
L’examen anatomopathologique montre un infiltrat lymphocytaire périvasculaire et périannexiel sans atypies cellulaires.
Les immunomarquages mettent en évidence la nature exclusivement T des lymphocytes.
Ce tableau bien individualisé cliniquement et histologiquement pose peu de problèmes de diagnostic différentiel avec un lymphome.
L’efficacité du thalidomide a été démontrée par une étude prospective randomisée en double insu.
5- Angiokératome acral pseudolymphomateux :
L’angiokératome acral pseudolymphomateux est une entité de description récente.
Une dizaine de cas ont été rapportés chez des enfants de 2 à 16 ans.
Il s’agit d’une éruption unilatérale de multiples papules de 1 à 5mm de diamètre, de couleur rouge ou violacée, angiomateuses, parfois kératosiques, situées sur les mains et les pieds.
Un cas de l’adulte a été rapporté, se présentant sous la forme d’une lésion unique du dos.
Il pourrait s’agir d’une variante de lésions induites par des piqûres d’insectes.
Pseudolymphomes cutanés B :
Des descriptions de pseudolymphomes cutanés B ont été rapportées sous diverses appellations : lymphocytome cutané bénin, lymphocytoma cutis, lymphadenosis benigna cutis, pseudolymphome de Spiegler-Fendt, ou hyperplasie lymphoïde bénigne.
De nombreuses étiologies ont été reconnues à l’origine des pseudolymphomes cutanés B, mais les cas pour lesquels aucune cause ne peut être déterminée sont les plus fréquents.
A – LYMPHOCYTOME BORRÉLIEN :
C’est une manifestation rare de l’infection par Borrelia burgdorferi.
Le lymphocytome borrélien est plus fréquent chez l’enfant.
Les lésions surviennent à la suite de piqûres de tiques en zone d’endémie borrélienne, après une période variant de quelques semaines à 10 mois.
Ce sont des papules ou des nodules érythémateux, uniques ou multiples, parfois associés à un érythème chronique migrant.
Par rapport à l’érythème chronique migrant, les lésions de lymphocytome sont plus tardives et plus prolongées.
Elles sont habituellement situées sur le visage ou le tronc.
Le lobe de l’oreille, l’aréole mammaire et le scrotum sont des localisations évocatrices, en rapport avec le tropisme du spirochète. Une adénopathie satellite est souvent détectée.
Les lésions ont une évolution chronique.
La sérologie borrélienne est positive dans environ la moitié des cas.
Le spirochète peut être mis en évidence dans les lésions par réaction de polymérisation en chaîne.
Le traitement repose sur l’administration de doxycycline ou d’amoxicilline pendant 14 jours, ou d’azithromycine pendant 5 jours.
Un début de régression des lésions est rapidement observé mais la régression complète peut nécessiter plusieurs semaines.
B – LYMPHOCYTOME SECONDAIRE À DES TATOUAGES, INJECTIONS, PIQÛRES D’INSECTES OU ZONA :
Un pseudolymphome cutané B peut être secondaire à des piqûres d’insectes, des injections réalisées pour des vaccinations ou des protocoles de désensibilisation, ou des réactions induites par le percement des lobes des oreilles avec mise en place d’un matériel métallique.
Les lésions sont des papules et des nodules érythémateux localisés aux sites d’injection.
Les pseudolymphomes B secondaires à des tatouages surviennent surtout dans les parties de couleur rouge, plus rarement dans les parties de couleur bleue ou verte.
Dans ces cas, les lésions histologiques comportent souvent des granulomes histiocytaires.
Des lésions de pseudolymphome B ont également été rapportées au site de lésions de zona.
C – PSEUDOLYMPHOMES MÉDICAMENTEUX :
Les pseudolymphomes médicamenteux ont habituellement un phénotype T mais quelques cas de pseudolymphomes B ont également été rapportés.
Ils réalisent alors des papules, des nodules ou des tumeurs asymptomatiques, uniques ou multiples.
L’examen histologique montre un infiltrat nodulaire constitué d’une majorité de lymphocytes B polytypiques. Les lésions régressent en quelques semaines à l’arrêt du médicament.
D – ANGIOKÉRATOME ACRAL PSEUDOLYMPHOMATEUX :
Parmi les quelques cas rapportés d’angiokératome acral pseudolymphomateux, une minorité a un phénotype B prédominant.
E – PSEUDOLYMPHOME B IDIOPATHIQUE :
C’est le plus fréquent des pseudolymphomes B cutanés.
Il réalise des papules, des nodules ou des tumeurs, de couleur rose ou rouge, asymptomatiques.
Les lésions sont en règle situées sur la tête ou le tronc et ont une évolution chronique.
Elles sont le plus souvent uniques et localisées, mais parfois multiples, groupées ou disséminées.
Il n’y a pas d’adénopathies, le reste de l’examen clinique est normal.
Aucune étiologie n’est retrouvée à l’interrogatoire.
L’examen anatomopathologique montre un infiltrat nodulaire ou diffus du derme, séparé de l’épiderme par une bande de tissu sain, constitué de lymphocytes associés à des quantités variables d’histiocytes, de polynucléaires éosinophiles et de plasmocytes.
Cet infiltrat peut être limité au derme papillaire ou s’étendre à tout le derme jusqu’à l’hypoderme.
Des aspects de centres germinatifs sont fréquents, associant des petits lymphocytes et des lymphocytes de plus grande taille.
Il existe également des macrophages et des cellules dendritiques.
Le lymphocytome à grandes cellules est caractérisé par un infiltrat constitué de cellules de grande taille avec quelques immunoblastes, entourées de petits lymphocytes.
L’infiltrat est polymorphe, associant des éosinophiles, des plasmocytes, des histiocytes et des cellules géantes.
Certains cas posent un problème de diagnostic différentiel très difficile avec un immunocytome.
Les immunomarquages montrent une prédominance de lymphocytes B polytypiques, associés à une quantité variable de lymphocytes T.
La recherche d’un clone B en biologie moléculaire est le plus souvent négative.
Dans certains cas, les lésions ont toutes les caractéristiques cliniques, anatomopathologiques et immunohistochimiques d’un pseudolymphome cutané bénin, mais la biologie moléculaire met en évidence la présence d’un clone B dans les lésions.
Cet élément ne doit pas remettre en cause le diagnostic, qui est porté sur la confrontation des éléments cliniques, histologiques, immunohistochimiques et évolutifs.
Il incite néanmoins à instituer chez ces patients une surveillance clinique prolongée.
En effet, si certains de ces cas restent au cours des années d’authentiques pseudolymphomes, d’autres évoluent vers un tableau de lymphome B cutané.
Dans ces cas, il s’agissait vraisemblablement dès le début d’un lymphome B de bas grade de malignité dont le diagnostic histologique et immunohistochimique était impossible en raison du caractère très minoritaire de l’infiltrat tumoral et/ou de l’importance quantitative des cellules réactionnelles.
Les lésions ont une évolution chronique.
Elles peuvent régresser spontanément ou récidiver après plusieurs mois ou années.
Elles peuvent être traitées par corticoïdes locaux ou intralésionnels, exérèse chirurgicale, thalidomide, interféron alpha, cryothérapie ou radiothérapie.
Diagnostic positif et diagnostic différentiel :
Le diagnostic de pseudolymphome et le diagnostic différentiel avec un lymphome peuvent être très difficiles.
Le diagnostic est porté sur une combinaison d’arguments cliniques, histologiques, immunophénotypiques, génotypiques et évolutifs.
A – TABLEAU CLINIQUE :
Les tableaux cliniques réalisés par les pseudolymphomes et les lymphomes cutanés peuvent être impossibles à différencier.
La mise en évidence d’une étiologie à l’interrogatoire peut être un argument en faveur du diagnostic de pseudolymphome.
Des lésions de grande taille, ulcérées, d’évolution rapide, ou la présence d’adénopathies font plutôt évoquer un diagnostic de lymphome.
En dehors de la papulose lymphomatoïde, les lésions multiples, disséminées et récidivantes sont également en faveur d’un lymphome.
Le contraste entre quelques lésions cliniques papuleuses ou papulonodulaires et une image histologique évocatrice de lymphome caractérise souvent les pseudolymphomes cutanés T.
Une lésion unique, de petite taille et une évolution indolente ou spontanément régressive peuvent se voir aussi bien dans les pseudolymphomes que dans certains cas de lymphomes.
B – EXAMEN ANATOMOPATHOLOGIQUE :
C’est un élément majeur du diagnostic de pseudolymphome et du diagnostic différentiel avec un lymphome.
Il importe donc de réaliser un examen anatomopathologique de taille suffisante et de bonne qualité. Les aspects histologiques réalisés par les pseudolymphomes et les lymphomes peuvent être très proches.
Néanmoins, certains éléments plaident en faveur de l’un ou l’autre de ces diagnostics.
Dans les infiltrats lymphocytaires cutanés à prédominance T, les signes histologiques suivants sont évocateurs du diagnostic de lymphome :
– alignement de lymphocytes dans la couche basale de l’épiderme ;
– halos clairs autour des lymphocytes situés dans l’épiderme ;
– taille plus grande et atypies des lymphocytes de l’épiderme ;
– groupements de lymphocytes dans l’épiderme ;
– absence de spongiose dans l’épiderme ;
– faisceaux de collagène épaissis dans le derme papillaire. Dans les lymphomes T cutanés, les thèques intraépidermiques sont constituées de lymphocytes à noyau cérébriforme, alors que dans les pseudolymphomes, les lymphocytes intraépidermiques sont accompagnés de plasmocytes et d’histiocytes.
Dans les infiltrats lymphocytaires cutanés à prédominance B, certains signes histologiques sont en faveur du diagnostic de pseudolymphome :
– l’hyperacanthose ;
– le polymorphisme de l’infiltrat ;
– la présence de cellules géantes ;
– le respect des annexes ;
– une prolifération vasculaire.
À l’inverse, les éléments en faveur du diagnostic de lymphome sont :
– l’absence de signes épidermiques ;
– des nappes de plasmocytes ;
– une infiltration diffuse du derme ou une infiltration de l’hypoderme ;
– la présence de corps de Dutcher (inclusions intranucléaires ou intracellulaires positives à l’acide périodique Schiff [PAS]).
Les centres germinatifs sont fréquents dans les pseudolymphomes, mais peuvent également être trouvés dans la partie réactionnelle de l’infiltrat d’un authentique lymphome.
C – EXAMENS IMMUNOHISTOCHIMIQUES :
Dans les pseudolymphomes T cutanés, la majorité des lymphocytes de l’infiltrat a un phénotype de lymphocytes T matures CD3+.
Contrairement aux lymphomes T cutanés qui ont un phénotype CD4+ prédominant, les pseudolymphomes cutanés T sont constitués d’un mélange de lymphocytes CD4+ et CD8+, et il n’y a pas de perte d’expression d’antigènes pan-T (CD2, CD3, CD5, CD7).
Dans le pseudolymphome actinique et le pseudolymphome lié à une infection par le VIH, le phénotype est majoritairement CD8+.
Dans les pseudolymphomes B cutanés, les immunomarquages montrent une majorité de lymphocytes exprimant les antigènes associés aux lymphocytes B (CD20), associée à une quantité variable de lymphocytes T.
L’élément le plus important pour le diagnostic est le caractère polytypique des lymphocytes B, qui sont constitués d’un mélange de lymphocytes exprimant la chaîne légère kappa et de lymphocytes exprimant la chaîne légère lambda.
Cet aspect s’oppose au caractère monotypique des lymphocytes de lymphome B cutané, c’est-à-dire qu’ils expriment tous le même type de chaîne légère.
Les immunomarquages représentent donc un élément important de diagnostic différentiel entre lymphomes et pseudolymphomes B cutanés.
Le diagnostic différentiel peut néanmoins être difficile avec certains lymphomes B où la population tumorale est minoritaire et où l’infiltrat T réactionnel est quantitativement important.
D – ÉTUDES GÉNOTYPIQUES :
Dans les pseudolymphomes, la recherche d’un clone T ou B en biologie moléculaire est le plus souvent négative.
Cette négativité peut constituer un argument de diagnostic différentiel avec les lymphomes T ou B, dans lesquels un clone est mis en évidence dans la majorité des cas. Cette distinction n’est cependant pas absolue.
Dans les infiltrats lymphocytaires à prédominance T, la biologie moléculaire représente la seule possibilité de démontrer le caractère monoclonal de l’infiltrat.
Cependant, la mise en évidence d’un clone n’est pas synonyme d’évolution maligne puisqu’un clone T est le plus souvent mis en évidence dans les lésions de papulose lymphomatoïde, qui ont une évolution bénigne.
Dans les pseudolymphomes cutanés B, un clone B peut être détecté dans une minorité de cas, avec une évolution prolongée qui reste bénigne. Cependant, certains cas ont évolué vers d’authentiques lymphomes B.
De même, il existe des cas de pseudolymphomes T avec présence d’un clone T cutané, qui ont évolué en quelques mois vers un véritable lymphome T.
Dans ces cas, il s’agissait probablement dès le début de lymphomes dont le diagnostic était impossible sur les examens anatomopathologiques et immunohistochimiques.
Conclusion :
Ainsi, dans les pseudolymphomes cutanés, les résultats de la biologie moléculaire peuvent constituer un élément supplémentaire pour le diagnostic, mais les données doivent toujours être interprétées en confrontant les éléments de la clinique, de l’examen anatomopathologique, des immunomarquages et de l’évolution.
Les études de réarrangement de gènes ne constituent pas un critère absolu permettant de différencier les pseudolymphomes des lymphomes vrais.
La découverte d’un clone T ou B ne doit pas faire modifier le diagnostic, mais inciter à une surveillance prolongée du patient.


")




")



{kind=link}