



















Introduction :
Les histiocytoses sont un groupe hétérogène de pathologies définies par la présence anormale dans les tissus de cellules ayant des caractéristiques communes (cellules non cohésives, noyau réniforme, cytoplasme abondant, chromatine différenciée) et qui sont soit des macrophages tissulaires, soit des cellules dendritiques.
La physiopathologie est assez polymorphe et variable selon la situation pathologique.
Dans certains cas, les histiocytes sont des macrophages exerçant leur fonction normale de phagocytose de substances biologiques produites ou non éliminées au cours d’erreurs innées du métabolisme (maladie de Gaucher, maladie de Fabry, maladie de Farber, maladies de Niemann-Pick).
Dans d’autres cas, la présence de ces cellules ne semble pas correspondre à une fonction donnée (histiocytoses primitives idiopathiques) comme dans la maladie d’Erdheim-Chester.
Histiocytoses de surcharge héréditaires (dyslipidoses) :
A – MALADIE DE GAUCHER :
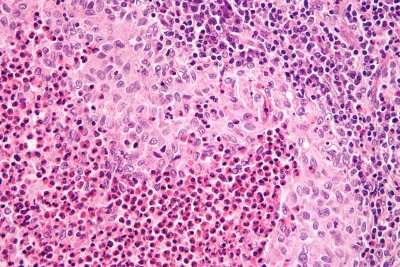 La maladie de Gaucher est la plus fréquente des affections héréditaires du métabolisme des glycolipides.
La maladie de Gaucher est la plus fréquente des affections héréditaires du métabolisme des glycolipides.
Il s’agit d’une maladie génétique autosomique récessive due au déficit d’une enzyme lysosomale, la bêta-glucocérébrosidase.
Il s’ensuit une accumulation pathologique de glucosylcéramide principalement dans le foie, la rate et la moelle osseuse.
Les manifestations cliniques sont essentiellement hépatospléniques et osseuses ; les manifestations ostéoarticulaires sont fréquemment révélatrices et constituent les principales sources de morbidité et d’invalidité.
1- Épidémiologie :
L’incidence de la maladie de Gaucher est difficile à évaluer car la présentation de la maladie est très hétérogène, comprenant tous les intermédiaires entre des formes totalement asymptomatiques et des formes très graves.
Elle est comprise entre 1/30 000 et 1/50 000 dans la population générale, plus élevée chez les juifs ashkénazes puisqu’elle est évaluée à 1/850.
Le registre mondial de la maladie de Gaucher publié en 2000 regroupe 1 698 patients.
2- Pathogénie :
Le déficit d’activité de la bêta-glucocérébrosidase, enzyme permettant d’hydrolyser le glucosylcéramide en céramide et glucose, conduit à une accumulation cellulaire de glucosylcéramide qui provient de la dégradation de la membrane cellulaire des hématies et des leucocytes ; dans le système nerveux central, il provient de la dégradation des gangliosides membranaires ; il s’accumule dans les lysosomes des macrophages, leur conférant l’aspect particulier des cellules de Gaucher.
Dans de rares cas, il a été trouvé un déficit d’un cofacteur de la bêta-glucocérébrosidase, la saposine.
Les lésions osseuses et viscérales peuvent être dues à des compressions vasculaires locales, une activation des macrophages par le biais d’une augmentation de production de cytokines, une synthèse accrue de catabolites toxiques du glucosylcéramide rendant compte de l’atteinte neurologique et une diminution de l’apoptose.
3- Génétique :
La maladie de Gaucher est une maladie génétique à transmission autosomique récessive.
Le gène de la bêta-glucocérébrosidase est localisé en 1q21.
Les cinq mutations les plus fréquentes sont N370S, 84GG, L444P, IVS2 et Rec (réarrangement entre le gène et le pseudogène).
* Différents phénotypes :
Le type 1 concerne la majorité des patients (94 %) adultes, le plus souvent juifs ashkhénases, avec une grande hétérogénéité dans l’expression clinique, allant de formes parfaitement asymptomatiques à des expressions viscérales graves, handicapantes, parfois mortelles, sans atteinte neurologique en dehors de syndromes parkinsoniens récemment décrits. Le type 2 concerne moins de 1 % des patients.
C’est la forme la plus sévère et la plus rare, débutant avant 6 mois, comportant une atteinte respiratoire, une hépatosplénomégalie et un syndrome neurologique sévère précoce (hypertonie axiale, trismus, strabisme, convulsions).
Les traitements sont inefficaces et le décès survient généralement avant 2 ans.
Le type 3 concerne 5 % des patients, caractérisé par une atteinte neurologique d’apparition plus tardive, moins sévère que dans le type 2.
Il est divisé en trois sous-types.
* Relations entre le phénotype et le génotype :
Il existe une relation entre les principales mutations connues du gène de la bêta-glucocérébrosidase et l’expression phénotypique de la maladie.
La présence d’un allèle N370S exclut une atteinte neurologique en dehors des syndromes parkinsoniens.
À l’inverse, la présence de L444P/L444P et D409H/D409H est associée de façon très forte au développement d’une forme neurologique au cours de la vie.
Les malades porteurs de L444P/L444P ont des degrés d’atteinte viscérale variables, mais ne développent jamais de calcifications valvulaires cardiaques, de cataracte ou d’hydrocéphalie associées au génotype D409H/D409H.
La connaissance du génotype ne permet pas cependant de prédire avec certitude le phénotype (type 1, 2 ou 3).
Les familles à risque doivent bénéficier d’un conseil génétique.
Des possibilités de diagnostic anténatal existent à partir du dosage de la bêta-glucocérébrosidase à partir de cellules amniotiques, du tissu des villosités du chorion, ou de la recherche de mutations connues dans la famille ou à implication phénotypique forte.
4- Manifestations osseuses :
Les manifestions osseuses sont volontiers révélatrices de la maladie et une atteinte osseuse est présente dans 80 % des types 1 et 3, constituant la principale cause de morbidité et d’invalidité.
On observe des douleurs osseuses dans 60 % des cas et articulaires dans 60 %, des ostéonécroses dans 57 % et des infarctus osseux dans 40 %.
* Infiltration médullaire :
Le tissu osseux est infiltré par les cellules de Gaucher qui entraînent une érosion endostale et un élargissement de la cavité médullaire ; les phénomènes ischémiques contribueraient également à l’ostéopénie trabéculaire.
L’infiltration médullaire se traduit par des douleurs rachidiennes et/ou articulaires, et par une hypertransparence radiologique à type d’images lytiques, hétérogènes avec des zones d’ostéosclérose, volontiers bilatérales.
* Déformations osseuses (remodelage osseux insuffisant) :
La diminution de l’activité ostéoclastique entraîne une diminution du modelage et du remodelage des os jeunes aboutissant à des déformations de la partie distale du fémur ou proximale du tibia dite en « flacon d’Erlenmeyer », généralement de façon bilatérale et symétrique, avec une perte de la concavité de la région métaphysaire et un élargissement anormal de l’os à cet endroit.
Ce signe n’est pas pathognomonique et peut s’observer dans la maladie de Pyle, la maladie de Niemann-Pick, la dysplasie craniométaphysaire et l’ostéopétrose.
Cette anomalie est asymptomatique, découverte de manière fortuite ou lors d’un bilan d’extension.
Il peut exister des déformations des corps vertébraux avec un affaissement des plateaux supérieurs et inférieurs, réalisant un aspect de « vertèbre en H » ; cette déformation peut se rencontrer dans certaines hémoglobinopathies.
Il pourrait s’agir soit d’un défaut de croissance à la jonction chondro-osseuse par atteinte vasculaire, soit d’un défaut de réparation d’un tassement précoce au cours de la croissance.
* Ostéopénie et fractures :
La diminution du nombre et de l’activité des ostéoprogéniteurs conduit à une diminution de la formation osseuse, et donc à une ostéopénie et à un risque accru de fractures.
La densité minérale osseuse de l’os trabéculaire et de l’os cortical est diminuée ; les marqueurs de résorption comme la pyridinoline et la désoxypyridoline urinaires sont augmentés, alors que les marqueurs de formation comme l’ostéocalcine ou les phosphatases alcalines osseuses restent dans des valeurs normales.
Les fractures vertébrales sont fréquentes (de 11 à 58 % des cas), chez les adultes mais aussi chez les enfants, avec quelques cas de compression médullaire.
Les fractures des os longs sont plus rares.
Il peut exister une érosion endostale bien visible sur certains os longs, notamment aux fémurs et aux tibias.
* Ostéonécroses et infarctus osseux :
L’ostéonécrose constitue la lésion osseuse la plus classique de la maladie de Gaucher.
Sa fréquence est de l’ordre de 50 % dans le type 1.
Les lésions d’ostéonécrose aseptique dans la maladie de Gaucher sont volontiers multiples et touchent les têtes humérales et fémorales, les condyles fémoraux et les plateaux tibiaux.
L’ostéonécrose médullaire se caractérise par des zones intramédullaires de densité élevée prédominant sur le bassin, et en particulier sur la sacro-iliaque.
Les infarctus osseux se manifestent le plus souvent par une symptomatologie douloureuse importante, siégeant au bassin ou sur un os long, avec des signes inflammatoires locaux, une réaction fébrile faisant craindre une complication infectieuse et pouvant dans certains cas révéler la maladie.
Les symptômes peuvent durer de plusieurs jours à plusieurs semaines.
Dans la corticale, l’infarctus osseux peut aboutir à une formation d’os nouveau intracortical donnant un aspect dit d’« os dans l’os ».
La splénectomie serait un facteur de risque important d’ostéonécrose du fémur, du tibia et de l’humérus : 14/15 (93 %) versus 21/36 (58 %) dans une série de 51 patients atteints de maladie de Gaucher avec ou sans ostéonécrose.
Le sexe masculin est un facteur de risque d’ostéonécrose humérale et du fémur distal.
* Infections ostéoarticulaires :
Il est classiquement décrit une augmentation de la fréquence des ostéomyélites dans la maladie de Gaucher dont la symptomatologie initiale n’est pas forcément facile à distinguer des « crises osseuses » ; en pratique, il semble qu’elles soient très rares et qu’elles aient été surévaluées en raison de l’allure très inflammatoire de ces « crises osseuses ».
Elles sont favorisées par la pratique d’une ponction-biopsie osseuse à visée diagnostique, qui doit donc être évitée en raison de son risque iatrogène potentiel sur ce terrain.
De ce fait, il faut utiliser au maximum les prélèvements bactériens périphériques (hémocultures…), des scintigraphies répétées, la tomodensitométrie ou l’imagerie par résonance magnétique (IRM).
Un germe atypique est souvent en cause et il faut en particulier rechercher des germes anaérobies. Les arthrites septiques sont rares.
5- Atteintes rares :
Des atteintes plus rares ont été décrites, à type de polyarthrite migratrice, d’atteintes des sacro-iliaques ou de la symphyse pubienne, de discopathies destructrices.
6- Imagerie à visée osseuse :
L’imagerie conventionnelle suffit souvent à évoquer le diagnostic.
Le problème vient de la possibilité d’aspects radiographiques similaires dans d’autres maladies, notamment dans les hémoglobinopathies et en particulier la drépanocytose.
La scintigraphie osseuse au 99mTechnétium permet une cartographie des différentes localisations osseuses de la maladie, notamment au début de la prise en charge.
Il s’agit en général d’hyperfixations ou d’hypofixations à la phase toute initiale d’un infarctus osseux.
La tomodensitométrie peut avoir un intérêt pour préciser une atteinte articulaire ou un envahissement des parties molles en cas d’infection.
L’IRM permet de voir précocement les lésions d’infarctus osseux ou d’ostéonécrose, les lésions infectieuses et les parties molles, mais également une modification du signal médullaire témoignant de l’infiltration pathologique.
On observe alors un signal de type « graisseux » avec un hypersignal en séquence T1 mais aussi T2.
Une corrélation entre l’activité médullaire et l’activité de la maladie a été montrée.
7- Atteintes non osseuses :
Une splénomégalie est palpée dans 95 % des cas, parfois responsable de douleurs liées à un infarctus splénique pouvant se compliquer de surinfection et engager le pronostic vital.
C’est l’hypersplénisme qui est au premier plan, avec une thrombopénie et parfois des saignements sévères.
Ces anomalies se corrigent après splénectomie mais récidivent.
En outre, la splénectomie favorise le développement de lésions d’autres organes ou l’aggravation de lésions préexistantes, notamment sur le plan osseux.
Une hépatomégalie est retrouvée dans 80 % des cas, aggravée par la splénectomie, rarement compliquée d’une cirrhose avec hypertension portale.
L’asthénie est fréquente, retentissant sur la qualité de vie mesurée sur l’échelle SF36 qui se normalise sous traitement.
L’atteinte pulmonaire peut avoir des causes extrinsèques comme une compression viscérale ou des fractures vertébrales et costales ; mais il existe des infiltrations spécifiques des capillaires et du parenchyme pouvant conduire à la fibrose et plus rarement à une hypertension artérielle pulmonaire.
La réponse au traitement enzymatique substitutif est variable.
Outre les manifestations neurologiques extrêmement sévères caractérisant les stades 2 et 3, des syndromes extrapyramidaux ont été décrits dans les formes de type 1, pouvant parfois précéder les manifestations de la maladie de Gaucher elle-même.
Les atteintes cardiaques, digestives, rénales et oculaires sont exceptionnelles.
Le risque d’hémopathie serait multiplié par 14,7 et de cancer solide par 3,6.
8- Diagnostic :
* Myélogramme ou biopsie ostéomédullaire :
Ils permettent de mettre en évidence des cellules de Gaucher, qui sont de grosses cellules macrophagiques dont le cytoplasme est plissé et qui contiennent des inclusions lysosomales constituées de glucosylcéramide.
Il existe de la bêta-glucocérébrosidase dans ces inclusions, mais elle n’est pas fonctionnelle.
Cependant, les cellules de Gaucher ne sont pas spécifiques de la maladie.
En effet, on peut retrouver des cellules avec les mêmes caractéristiques cytologiques (« pseudo-Gaucher ») dans la leucémie myéloïde chronique, au cours du myélome ou d’autres hémopathies.
Le myélogramme permet de caractériser le type de cytopénie.
* Dosage de la bêta-glucocérébrosidase :
Il permet de poser le diagnostic.
Il est fait sur leucocytes circulants, sur culture de fibroblastes cutanés, mais également sur cellules amniotiques ou trophoblastiques dans le cadre d’un diagnostic prénatal.
Les valeurs de l’activité enzymatique sont de 10 à 30 % des valeurs normales, parfois moins.
Le taux n’est pas prédictif de la sévérité de la maladie.
* Recherche de mutations :
On peut rechercher les cinq mutations les plus fréquentes par polymerase chain reaction, dont on a vu qu’elles avaient des liens avec l’expression clinique de la maladie.
L’absence d’une de ces cinq mutations ne doit pas faire écarter le diagnostic puisqu’on décompte environ 200 mutations différentes.
* Marqueurs biologiques :
Certains marqueurs biologiques sont le reflet de la thésaurismose.
Il s’agit en premier lieu des cytopénies qui découlent de la splénomégalie et du syndrome d’hypersplénisme qu’elle entraîne.
La plus habituelle est la thrombopénie, notamment chez les patients non splénectomisés.
Le myélogramme, outre les cellules de Gaucher qu’il peut mettre en évidence, permet d’en préciser le mécanisme, soit central par infiltration médullaire, soit périphérique par destruction ou séquestration splénique.
L’anémie est souvent au second plan.
Les neutropénies sont parfois sévères.
Il peut exister des troubles de la coagulation à type d’allongement du temps de céphaline activé, soit sans déficit en facteur de coagulation, dû à l’interférence des sphingolipides avec les protéines de la coagulation, soit lié à une diminution des facteurs de coagulation par consommation par les macrophages activés.
Les accidents hémorragiques sont rares en dehors des thrombopénies sévères.
Il existe une augmentation de l’enzyme de conversion de l’angiotensine, une hypergammaglobulinémie polyclonale (mais aussi parfois des gammapathies monoclonales de signification inconnue et des myélomes), une hyperferritinémie avec un fer sérique et un coefficient de saturation de la transferrine normaux, témoignant d’une activation de la fonction macrophagique.
L’augmentation des phosphatases acides tartrate-résistantes (isoenzyme 5b) et de la chitotriosidase (jusqu’à 600 fois les valeurs normales) reflète l’atteinte lysosomale.
Ces éléments sont de bons moyens de suivi de l’évolution de la maladie, notamment sous traitement.
Ils ne sont pas spécifiques puisqu’ils sont élevés dans d’autres affections lysosomales comme la maladie de Niemann-Pick, la sarcoïdose ou la leishmaniose viscérale. Dans 6 % de la population générale, la chitotriosidase est indosable du fait d’une mutation du gène.
La néoptérine, l’adénosine désaminase, la bêta-hexosaminidase sont également augmentées et il existe une bonne corrélation de toutes ces molécules, ainsi que de l’angiotensine convertase et de la chitotriosidase, avec le degré d’activation des monocytes/ macrophages, mais pas entre les molécules, ce qui suggère qu’elles représentent un aspect différent de cette activation.
Les lymphocytes T pourraient jouer un rôle régulateur de l’activité ostéoclastique.
Chez les patients ayant une atteinte osseuse, il a été observé des taux de lymphocytes TCD8 plus bas, corrélés à des taux plus élevés de phosphatases acides tartrate-résistantes, reflétant l’activité accrue des ostéoclastes.
L’augmentation du catabolisme du cholestérol (LDL et HDL) par les macrophages dans la maladie de Gaucher conduit à une diminution de la cholestérolémie.
Même les porteurs sains ont des taux de cholestérol plus bas, ce qui suggère un lien génétique entre le locus du gène de la bêta-glucocérébrosidase et celui de l’hypo-alipoprotéinémie familiale qui donne des coronaropathies précoces et dont le gène n’a pas été identifié.
9- Traitement :
* Traitement symptomatique :
La douleur, et tout particulièrement la douleur osseuse, doit être prise en charge par des antalgiques de niveau I et II, voire des morphiniques. Une immobilisation peut être utile.
La splénectomie a perdu beaucoup de ses indications (complications hématologiques) depuis que le traitement substitutif est disponible ; elle est actuellement réservée aux échecs de la thérapie substitutive et aux situations d’urgence.
Les traitements de l’appareil locomoteur et de son handicap, en dehors de la douleur, font appel aux arthroplasties, notamment en cas d’ostéonécrose.
Il faut prendre garde aux infections postopératoires, souvent à germes atypiques, et entourer le geste d’une antibiothérapie parfois prolongée active contre les bactéries à Gram positif 7 jours après l’intervention chirurgicale.
La raréfaction osseuse peut bénéficier dans les cas sévères de traitements par bisphosphonates.
Le pamidronate par voie intraveineuse a montré une efficacité, au moins sur la densité minérale osseuse.
* Traitement substitutif :
Il s’agit d’un changement considérable dans la prise en charge des malades et du premier traitement « étiologique » dans une thésaurismose.
En 1991, Barton et al. produisent une enzyme partiellement déglycosylée, dévoilant des résidus mannose, capable de se fixer à des récepteurs macrophagiques spécifiques et permettant son internalisation dans la cellule vers les lysosomes : c’est l’aglucérase (Cérédaset).
À partir de 1996, l’enzyme est produite à partir de cellules CHO : c’est l’imiglucérase (Cérézymet).
Il existe moins de risque potentiel de transmission infectieuse avec la nouvelle molécule.
La posologie utilisée est de 60 UI/kg/15 jours en perfusions de 2 à 3 heures.
Il est possible de diminuer les doses par la suite en fonction de la réponse clinique et biologique à 45 UI/15 jours puis 30 UI/15 jours.
En France, l’autorisation du traitement est soumise à l’approbation d’un comité national d’évaluation (Comité d’étude du traitement de la maladie de Gaucher [CETG]).
La raison en est le coût extrêmement élevé du médicament.
En mai 2003, le prix d’un flacon de 400 UI était de 1771,26 , soit pour un patient de 70 kg un coût annuel de 446 352 (2 936 264 F) !
Le traitement s’adresse aux patients ayant une forme symptomatique de maladie de Gaucher de type 1 et à l’ensemble des malades de type 3.
Il n’y a pas d’indication pour le type 2 car le traitement n’a pas d’influence sur l’évolution de la maladie neurologique.
La tolérance est généralement bonne ; de 10 à 15 % des patients développent des anticorps, mais il y a peu de réactions anaphylactiques ou de résistance au traitement.
La grossesse reste une contre-indication.
Le traitement est efficace sur l’asthénie, les douleurs osseuses et abdominales, l’hépatosplénomégalie. Les atteintes radiologiques mettent beaucoup plus de temps à régresser, quand elles régressent.
Les atteintes neurologiques et l’hypertension artérielle pulmonaire ne semblent pas répondre au traitement substitutif.
Les cytopénies et les marqueurs diminuent sous traitement mais récidivent à l’arrêt.
Il existe une amélioration de la qualité de vie.
* Thérapie génique :
Il s’agit d’introduire le gène de la bêta-glucocérébrosidase dans des cellules souches hématopoïétiques que l’on va réinjecter au patient, mais les taux de bêta-glucocérébrosidase restent encore trop faibles pour obtenir un effet clinique.
L’enjeu des recherches actuelles est de trouver des méthodes permettant de concentrer les cellules transfectées.
* Autres voies thérapeutiques :
Le traitement substitutif ne permet pas de résoudre tous les problèmes liés à la maladie, car il nécessite un traitement parentéral continu et n’est pas efficace sur certaines manifestations cliniques installées ou sur les formes neurologiques.
De plus, son coût est exorbitant.
Les allogreffes médullaires ont donné de bons résultats ; mais le risque thérapeutique étant majeur, elles ne méritent d’être tentées que dans les formes neurologiques graves.
Un essai avec une molécule dénommée OGT 918 (N-butyldéoxynojirimycine) ou miglustat a montré une efficacité modérée. Cette molécule diminue les taux de substrat de la bêta-glucocérébrosidase.
Elle a l’avantage d’une prise orale (100 mg trois fois par jour), mais a une efficacité moindre que le traitement substitutif et des effets secondaires fréquents (diarrhée, perte de poids, neuropathie périphérique).
Cependant, le miglustat (Zavescat) a obtenu une autorisation de mise sur le marché européenne en novembre 2002.
Son indication semble être réservée actuellement aux cas d’intolérance ou d’inefficacité du traitement substitutif.
B – MALADIE DE FABRY :
La maladie de Fabry est une sphingolipidose de transmission récessive liée au chromosome X, due à un déficit partiel ou complet de l’alpha-galactosidase, conduisant à l’accumulation de glycophospholipides dans les lysosomes des cellules endothéliales et musculaires lisses des vaisseaux, épithéliales de la cornée, des glomérules et tubules rénaux, dans les cardiomyocytes et les ganglions du système nerveux autonome.
Ce déficit métabolique entraîne une neuropathie douloureuse, des angiokératomes, une cardiomyopathie, une encéphalopathie ischémique avec accidents vasculaires cérébraux et une insuffisance rénale.
1- Formes cliniques :
Dans sa forme classique, la maladie atteint les hommes hémizygotes et débute dans l’enfance ou l’adolescence par des acroparesthésies et des angiokératomes suivis d’une atteinte oculaire, cardiorespiratoire, neurologique, digestive et rénale.
Les manifestations ostéoarticulaires comportent un gonflement périarticulaire des genoux, des coudes et des petites articulations distales des doigts qui sont déformées et limitées en extension, une ostéonécrose de la tête fémorale et de l’astragale, une raréfaction osseuse thoracolombaire.
L’activité alpha-galactosidase A est effondrée.
La forme atypique ne comporte pratiquement que des troubles cardiaques d’apparition tardive (hypertrophie ventriculaire, arythmie, atteinte valvulaire), en raison d’une activité résiduelle (de 5 à 35 %) de l’alpha-galactosidase.
Chez les femmes hétérozygotes, conductrices de la maladie, la symptomatologie clinique commence plus tardivement et reste le plus souvent modérée ; une dystrophie cornéenne asymptomatique est présente dans 70 % des cas, des angiokératomes modérés dans 30 % et une neuropathie douloureuse dans moins de 10 %.
Les atteintes viscérales sévères sont rares.
La détection de la mutation du gène GLA permet l’identification des conductrices.
Le diagnostic prénatal repose sur le dosage de l’activité enzymatique sur villosités choriales ou cellules amniotiques.
2- Traitement :
Le traitement comporte des mesures symptomatiques (douleurs), la dialyse et la transplantation pour les insuffisants rénaux terminaux.
Deux traitements substitutifs enzymatiques ont eu en 2001 une autorisation de mise sur le marché en Europe : l’agalsidase alpha (Replagalt) et l’agalsidase bêta (Fabrazymet).
3- Maladie de Farber :
La maladie de Farber (ou lipogranulomatose) est une affection rare et progressive de l’enfance, due à un déficit en céramidase acide lysosomale, responsable d’une accumulation intracellulaire de céramides dans les reins, le foie, les poumons, les ganglions lymphatiques et la peau.
Ce déficit entraîne un retard de croissance et se révèle au cours des premiers mois de la vie par des déformations articulaires douloureuses, des nodules périarticulaires et sous-cutanés multiples, une voix rauque et une insuffisance respiratoire.
Les nodules augmentent en taille et en nombre au cours de l’évolution, le plus souvent sur les interphalangiennes des mains, des poignets, des coudes et des chevilles ; ils peuvent se développer dans la conjonctive, les oreilles, le nez, l’occiput et le rachis.
Le décès survient dans les premières années de la vie.
Certaines formes sont d’apparition plus tardive.
L’histologie montre une infiltration des cartilages, des os et des tissus mous par des histiocytes et des macrophages contenant des corps de Farber dans leurs vacuoles, des cellules spumeuses au sein d’un granulome.
C – MALADIES DE NIEMANN-PICK :
Les maladies de Niemann-Pick sont aujourd’hui classées en deux catégories : celles dues au déficit primitif en sphingomyélinase acide (types A et B) et celle due au déficit d’une protéine (NPC1) régulant le transport intracellulaire du cholestérol-LDL (type C).
Une infiltration de cellules spumeuses et une organomégalie sont les caractères communs à cet ensemble de pathologies héréditaires.
Soulignons qu’une atteinte neurologique sévère survient uniquement dans les types A et C, et non dans le type B.
L’accumulation de lipides, et en particulier de sphingomyéline, peut être trouvée dans les cellules réticuloendothéliales et parenchymateuses de nombreux tissus, les gouttelettes lipidiques incluses dans le cytoplasme des macrophages prenant une couleur « bleu de mer » après coloration de May-Grünwald-Giemsa.
1- Manifestations cliniques :
Les malades de type A ont un taux résiduel de sphingomyélinase de moins de 5 % ; l’affection débute tôt dans l’enfance et se traduit par une hépatomégalie et une atteinte neurologique conduisant au décès dans les 3 premières années de la vie.
Les malades de type B ont un taux résiduel de sphingomyélinase de 5 à 10 % ; il existe aussi une hépatosplénomégalie facilement repérable dans l’enfance, mais il n’y a pas d’atteinte neurologique, ce qui permet la survie à l’âge adulte.
C’est la présence ou non d’une infiltration pulmonaire qui détermine le pronostic dans ces formes tardives.
Les malades de type C ont des manifestations cliniques plus hétérogènes, avec une hépatosplénomégalie variable mais des atteintes neurologiques sévères (hypotonie, ataxie, dystonie, retard mental) conduisant à un décès dans la première décennie pour la forme infantile, dans la deuxième décennie pour la forme juvénile.
Le début à l’âge adulte est rare.
2- Manifestations ostéoarticulaires :
Les cellules spumeuses s’accumulent dans la moelle osseuse et modifient les travées osseuses, de façon relativement fréquente dans le type B où la survie est prolongée.
Dans les os longs, la corticale est amincie ; la taille et le nombre des travées de l’os spongieux sont diminués, entraînant une hypertransparence osseuse et un élargissement de la médullaire que l’on retrouve dans la maladie de Gaucher ; l’infiltration de la moelle osseuse des os longs peut être précisée par l’IRM.
Les mêmes modifications peuvent toucher les os tubulaires des mains, dans les types B et C.
Une déformation en coxa valga est fréquente dans les types A et C.
Des fractures vertébrales ont été décrites dans le type B.
L’ossification de nombreux noyaux secondaires peut être retardée.
Il n’y a pas cependant de lésions lytiques ni d’ostéonécrose comme dans la maladie de Gaucher. Le seul traitement spécifique est la greffe de moelle osseuse.
La thérapie enzymatique substitutive n’est pas encore disponible.
Histiocytoses sporadiques :
A – MALADIE DE CHESTER-ERDHEIM :
La maladie de Chester-Erdheim fait partie du cadre nosologique des histiocytoses non langerhansiennes.
Un cardiologue américain Chester et un anatomopathologiste viennois Erdheim ont les premiers décrit en 1930 une forme de granulomatose lipoïdique distincte des autres maladies histiocytaires.
À ce jour, plus d’une centaine de cas ont été publiés, le plus souvent sous forme d’observations isolées.
La plus importante revue de la littérature a été réalisée en 1996 par Veyssier-Bellot et al. qui ont colligé 59 cas, dont sept personnels.
La maladie a été observée à tout âge (de 7 à 84 ans) et sans prédominance selon le sexe.
1- Manifestations cliniques :
* Atteinte osseuse :
Les douleurs osseuses sont présentes chez près de la moitié des patients et représentent le signe clinique le plus fréquent.
Elles touchent préférentiellement les membres inférieurs, et particulièrement les genoux, les tibias et les chevilles.
Elles peuvent s’accompagner d’une augmentation de la chaleur locale, voire d’un gonflement et d’une rougeur. Parfois permanentes, elles peuvent aussi évoluer par poussées.
Les radiographies standard mettent en évidence une ostéosclérose bilatérale et symétrique des os longs, médullaire et corticale, touchant principalement les régions diaphysaires et métaphysaires.
Les os longs le plus fréquemment atteints sont les fémurs, les tibias, les péronés, les cubitus, les radius et les humérus.
Plus rarement, des zones d’ostéolyse sont associées sur les os longs et parfois les os plats. Le squelette axial est beaucoup plus rarement atteint.
La scintigraphie osseuse révèle une hyperfixation symétrique des os longs ; elle permet une évaluation de l’extension des lésions osseuses et le suivi des lésions, en particulier après traitement.
L’IRM montre une diminution du signal dans les séquences pondérées en T1 correspondant au remplacement de la moelle osseuse par l’infiltration lipidique.
Sur les séquences pondérées en T2, il existe des zones d’hyposignal ou d’hypersignal selon l’intensité de l’ostéosclérose.
* Atteintes extraosseuses :
Elles sont rapportées dans près de la moitié des cas et sont très variées.
L’atteinte rétropéritonéale est le plus souvent asymptomatique.
Plus rarement, elle est responsable d’une hydronéphrose et de douleurs abdominales.
La tomodensitométrie permet de mettre en évidence les dépôts xanthogranulomateux rétropéritonéaux.
Les lésions peuvent toucher également les reins par envahissement périrénal.
L’exophtalmie est liée aux dépôts xanthogranulomateux rétroorbitaires et fait courir le risque de cécité.
Elle peut être révélatrice de la maladie.
Elle est le plus souvent bilatérale, symétrique et douloureuse.
Elle est bien mise en évidence par la tomodensitométrie et l’IRM qui montrent l’envahissement orbitaire avec atteinte des nerfs optiques et des muscles oculomoteurs.
Les manifestations endocriniennes sont dominées par le diabète insipide.
Plus rarement peuvent être observées une hyperprolactinémie et une insuffisance gonadotrope.
Les localisations cutanées se résument à des xanthomes plans touchant le plus souvent les paupières mais pouvant également atteindre le tronc.
Les manifestations neurologiques sont protéiformes, présentes dans un tiers des cas, essentiellement à type de syndrome cérébelleux, plus rarement à type de compression médullaire.
L’IRM permet de confirmer les localisations cérébrales symptomatiques, de dépister des localisations infracliniques et d’apprécier l’atteinte épidurale.
Elle montre des signaux hyperintenses sur les séquences en T2 avec rehaussement après injection de gadolinium.
Des dépôts le long de la dure-mère, de la faux du cerveau ainsi que de la tente du cervelet peuvent être ainsi détectés.
Le liquide céphalorachidien est le plus souvent normal ; il existe parfois une hyperprotéinorachie.
Les manifestations pulmonaires sont de type bronchique, pulmonaire ou pleural.
La radiographie standard montre un aspect de pneumopathie interstitielle prédominant aux sommets.
Sur la tomodensitométrie, il existe des opacités réticulaires et centrolobulaires, et des zones avec aspect de verre dépoli. Un épaississement de la plèvre viscérale peut être associé.
La granulomatose lipidique peut toucher plus rarement le péricarde, les muscles et la glande mammaire.
* Signes généraux :
Ils sont présents dans 18 % des cas.
Ils sont le plus souvent associés aux formes avec atteinte extraosseuse et sont constitués par une altération de l’état général (asthénie, amaigrissement) et de la fièvre.
2- Signes biologiques :
Il n’existe aucun signe biologique spécifique.
Le syndrome inflammatoire est l’anomalie le plus fréquemment rencontrée.
Le bilan lipidique est normal. Le bilan phosphocalcique est le plus souvent normal.
Une augmentation des phosphatases alcalines a parfois été rapportée.
Exceptionnellement, il peut exister une élévation de l’ostéocalcine.
3- Anatomie pathologique :
Les sites de biopsie le plus souvent utilisés sont l’os, les masses rétropéritonéales, les masses rétro-orbitaires et la peau.
L’examen histologique met en évidence une infiltration par des histiocytes spumeux riches en graisse associée à une fibrose.
À côté de ces histiocytes, il existe des cellules géantes multinucléées de type cellules de Touton.
En revanche, il n’y a pas d’infiltrat éosinophile.
L’immunohistochimie montre que les histiocytes sont très fortement positifs avec le marquage CD 68.
En revanche, ils sont le plus souvent négatifs avec les marquages CD 1a et S100.
Ces deux caractéristiques permettent de différencier la maladie de Chester-Erdheim de l’histiocytose langerhansienne.
De plus, l’examen en microscopie électronique ne retrouve jamais de granules de Bierbeck ni de corps X.
4- Pathogénie :
La pathogénie de la maladie de Chester-Erdheim n’est à ce jour pas parfaitement élucidée.
L’hypothèse d’une maladie métabolique responsable d’une anomalie de stockage des lipides est sous-tendue par l’étude biochimique des lésions qui montre une prédominance d’esters de cholestérol.
L’hypothèse d’une maladie néoplasique a été aussi soulevée devant la mise en évidence d’une prolifération clonale.
La recherche d’une clonalité s’est avérée positive chez trois patients en étudiant après extraction de l’acide désoxyribonucléique le gène du récepteur androgène humain (Humara) en polymerase chain reaction.
Cependant, cette recherche n’est pas constamment positive.
5- Frontière nosologique :
L’association de la maladie de Chester-Erdheim à une histiocytose langerhansienne a été rapportée à plusieurs reprises.
Cette association pourrait s’expliquer par deux hypothèses : soit l’intervention d’un stimulus commun induisant la prolifération indépendante des deux lignées cellulaires, soit une transformation d’une histiocytose langerhansienne en une maladie de Chester-Erdheim.
Cette deuxième hypothèse est sous-tendue par l’apparition possible à la phase tardive de l’histiocytose langerhansienne d’un processus fibrosant et d’une accumulation de lipophages avec la disparition éventuelle du marquage par le CD1a.
6- Pronostic :
Dans la série de 59 patients de Veyssier-Belot et al., 22 décès ont été observés dans un délai allant de 3 mois à 10 ans.
La sévérité du pronostic est réglée par l’existence de localisations extraosseuses.
Ce sont les localisations neurologiques, pulmonaires, cardiaques et rénales qui sont les plus graves.
Cependant, les formes initialement osseuses peuvent secondairement se compliquer de localisations viscérales.
7- Traitement :
Le traitement n’est absolument pas codifié. En cas de localisations osseuses douloureuses résistant aux antalgiques usuels, la radiothérapie a été préconisée.
Dans les cas avec atteintes viscérales, la corticothérapie utilisée seule s’est avérée le plus souvent inefficace.
La chimiothérapie par vinblastine, doxorubicine, étoposide ou méthotrexate a été efficace dans un faible nombre de cas. Enfin, l’intérêt du traitement par interféron alpha a été décrit.
B – RÉTICULOHISTIOCYTOSE MULTICENTRIQUE (RHM) :
La RHM est une maladie systémique rare, d’étiologie inconnue, caractérisée par une atteinte cutanée et une polyarthrite destructrice.
Elle est liée à une accumulation d’histiocytes et de cellules géantes dans la peau, dans les articulations et parfois dans l’os.
Elle entre dans le cadre d’un syndrome paranéoplasique dans un quart des cas.
Elle peut être associée à la tuberculose ou à des pathologies auto-immunes.
Cette pathologie est décrite sous plusieurs vocables : réticulohistiocytose, dermatoarthrite lipoïdique, xanthomatose cutanée normolipidémique, histiocytose à cellules géantes.
Mais la dénomination RHM est maintenant la plus utilisée.
La première description de RHM a été rapportée il y a plus de deux siècles, puis précisée en 1969 par Barrow et Holubar avec une série de 33 cas.
Plus de 200 cas ont été rapportés dans la littérature, le plus souvent sous forme de cas isolés.
1- Épidémiologie :
Cette affection est rare.
Elle touche le plus souvent les femmes avec un sex-ratio de 2/3.
Il n’a pas été retrouvé de prédisposition familiale.
Elle peut survenir à tout âge (forme pédiatrique et sénile), et particulièrement entre 40 et 50 ans.
L’incidence est faible mais probablement sous-estimée par la méconnaissance de cette pathologie et sa confusion avec d’autres polyarthrites.
2- Manifestations cliniques :
Le début de la maladie est insidieux.
Des signes généraux avec fièvre, amaigrissement, malaise sont décrits.
La RHM débute par des douleurs articulaires dans près de la moitié des cas.
Les signes cutanés inaugurent la maladie dans 18 % des cas et s’associent à la polyarthrite dans 21 % des cas.
* Manifestations articulaires :
La rapidité d’évolution et la sévérité de la polyarthrite sont deux des caractéristiques de la RHM.
C’est une polyarthrite destructrice, symétrique, avec atteinte préférentielle des articulations interphalangiennes distales.
L’aspect de « main en lorgnette » est caractéristique : mains aux doigts raccourcis, d’une consistance spongieuse.
La RHM peut toucher toutes les articulations, y compris la temporomandibulaire : articulations interphalangiennes (76 %), genoux (73 %), épaules (64 %), poignets (64 %), hanches (61 %), chevilles (58 %), pieds (58 %), coudes (58 %), vertèbres (55 %).
Des confusions avec la polyarthrite rhumatoïde sont fréquentes, surtout avant l’apparition de lésions cutanées évocatrices.
Mais il existe cliniquement des différences entre ces deux arthropathies : atteinte des interphalangiennes distales, atteinte du cartilage et de l’os sous-articulaire avec des déformations plus importantes, évolution classiquement plus sévère et plus rapide dans le cas de RHM.
La RHM peut être aussi confondue avec le rhumatisme psoriasique du fait de l’atteinte des interphalangiennes distales ou avec une arthropathie goutteuse avec des « pseudotophus ».
* Manifestations cutanéomuqueuses :
Elles consistent en de multiples lésions papulonodulaires prurigineuses, de couleur rouge violacé ou chamois, siégeant sur l’ensemble du corps, et particulièrement sur la face (94 %) et les mains (91 %).
L’atteinte digitale peut simuler un tableau de dermatomyosite.
L’évolution vers l’ulcération ou la nécrose est fréquente. Leur incidence diminue dans la direction céphalocaudale.
La distribution faciale de ces nodules est caractéristique.
Le cuir chevelu, en particulier en région rétroauriculaire, est souvent atteint, avec de nombreuses lésions confluant pour former des petites tumeurs.
À l’oreille, l’hélix est élargi, sensible et rouge.
Le nez est déformé par des masses nodulaires.
L’ensemble réalise un faciès léonin. Parallèlement, les muqueuses sont atteintes, avec des caractéristiques physiques similaires : lèvres (30 %), muqueuse buccale (15 %), septum nasal (12 %), langue (9 %), gencives (6 %).
Aucune atteinte génitale ou anale n’a été rapportée.
Les xanthélasmas sont présents dans un tiers des cas, sans association significative avec une hypercholestérolémie.
* Autres manifestations :
Les manifestations systémiques sont variées, avec une incidence difficile à apprécier en raison des formes asymptomatiques.
Elles résultent de l’infiltration des tissus par des cellules géantes et des histiocytes.
Le système musculaire peut être atteint, mimant dans certains cas des polymyosites.
Classiquement, l’atteinte est fruste, avec des myalgies et une faiblesse musculaire modérée.
Des observations de myopathies sévères et spécifiques ont été décrites, avec l’aspect de myopathie dégénérative multivacuolaire extensive à la biopsie.
Les manifestations viscérales sont essentiellement cardiovasculaires (péricardite, endocardite), pulmonaires (fibrose pulmonaire), plus rarement digestives.
Une forme purement cutanéoarticulaire peut évoluer avec le temps vers une forme systémique comme le montre l’observation de Ka et al. où une cardiopathie est apparue 20 ans après les premiers symptômes.
3- Manifestations paracliniques :
Aucun test biologique sérique spécifique n’a été mis en évidence.
Les radiographies orientent le diagnostic, mais c’est la biopsie cutanée ou synoviale qui l’affirme.
* Signes biologiques :
Il existe un syndrome inflammatoire dans certains cas, avec une anémie, une augmentation de la vitesse de sédimentation et de la protéine C réactive.
Des observations avec des facteurs rhumatoïdes positifs ont été rapportées.
En cas d’atteinte musculaire, la créatinine phosphokinase peut être augmentée.
Les dosages des triglycérides et du cholestérol ne sont pas modifiés de façon significative.
Le liquide synovial est d’aspect variable : mécanique, inflammatoire à prédominance de polynucléaires.
* Signes radiologiques :
Les radiographies standards des articulations orientent vers le diagnostic de RHM, avec des signes différents des autres arthropathies :
– érosions à bords nets, d’abord marginales puis centrales, le plus souvent symétriques, affectant spécialement les interphalangiennes distales et les métatarsophalangiennes, accompagnées d’un épaississement des parties molles (nodules) ;
– ostéolyse ponctuée des épiphyses de 1 mm à 1 cm ;
– absence ou discrétion de la déminéralisation osseuse, de la réaction périostée et d’ostéophytes, même en présence de grandes destructions articulaires (résorption des phalanges) ;
– atteinte précoce et sévère de l’articulation atloïdoaxoïdienne ;
– érosions de la sacro-iliaque, sans condensation.
* Anatomie pathologique :
En microscopie optique, la mise en évidence de cellules mononucléées ou multinucléées réalisant des cellules géantes avec un cytoplasme éosinophile PAS positives est caractéristique : aspect finement granuleux, opaque, aspect ground glass.
La forme et la taille de ces histiocytes sont irrégulières, avec un diamètre parfois de plus de 100 µm.
Les noyaux de ces cellules géantes sont nombreux, plus de 20 généralement.
Dans les cellules les plus jeunes, on trouve plutôt des infiltrats lymphocytaires et vasculaires denses.
Le développement de l’infiltrat refoule le collagène qui fait l’objet d’une phagocytose.
La microscopie électronique révèle un grand nombre de granules denses près de l’appareil de Golgi, phosphatase alcaline positifs, et entourés d’un halo suggérant la présence de lysosomes.
Aucun corps X ou granule de Bierbeck n’est retrouvé.
Les vacuoles lipidiques au sein des histiocytes reflètent un processus dégénératif, plutôt qu’une surcharge. Plusieurs immunomarquages sont positifs : CD4, CD45, CD68 (caractéristique de la lignée monocytes-macrophages), HLA-DR, lysosyme, a1 antitrypsine.
Classiquement, les immunomarquages CD20, S100 et le facteur XIIIa sont négatifs.
Pour chaque site (cutané, synovial, cardiaque…), cette description est valable.
4- Évolution. Pronostic :
L’évolution est chronique avec des poussées entrecoupées de rémissions.
Dans la plupart des cas, la RHM se stabilise avec ou sans traitement après quelques années d’évolution (8 ans en moyenne) avec des arthrites quiescentes et des lésions cutanéomuqueuses séquellaires. Récemment, Ka et al. ont rapporté un cas évoluant depuis 20 ans, compliqué d’une cardiomyopathie.
Grâce à l’utilisation d’immunosuppresseurs, les formes mutilantes responsables de handicap majeur et de défigurations définitives sont rares, de l’ordre de 10 %.
5- Pathologies associées :
Différentes pathologies sont associées à la RHM.
Dans 25 % des cas, elle s’intègre dans le cadre d’un syndrome paranéoplasique en précédant de quelques mois l’apparition d’un cancer.
La particularité des RHM dites paranéoplasiques est l’atteinte cutanée isolée dans 90 % des cas.
En pratique, la recherche systématique d’une néoplasie et d’une tuberculose (5 % des cas) est indispensable. Son association est possible avec certaines connectivites, en particulier le syndrome de Gougerot-Sjögren.
6- Traitement :
La nécessité d’une prise en charge dès les premiers symptômes est actuellement admise.
Cette attitude était initialement controversée car la RHM se stabilise spontanément en quelques années. Cependant, la rapidité de l’évolution, le risque de séquelles et de complications systémiques justifient un traitement agressif.
Différentes molécules ont été proposées avec efficacité : hydroxychloroquine, méthotrexate, ciclosporine, cyclophosphamide…
Plus récemment, l’efficacité d’un traitement par anti-tumor necrosis factor a été montrée. Liang et Granston ont proposé un schéma thérapeutique à partir d’une revue de 13 cas en association avec une corticothérapie : méthotrexate en première intention, cyclophosphamide ou chlorambucil en cas d’échec ou d’intolérance. Par ailleurs, le traitement efficace de la pathologie sous-jacente permet la régression de la RHM.







")



{kind=link}