



















Introduction :
Les dysplasies pilaires correspondent à l’ensemble des modifications de densité, de structure, de forme et de couleur affectant le cheveu, se manifestant dès l’enfance ou au cours des premiers mois.
À ce titre, le cheveu correspond à un marqueur d’un trouble génétique ou métabolique.
Son examen clinique ou par les examens paracliniques est donc primordial.
Méthodes d’exploration :
 Les tiges pilaires sont évidemment très accessibles aux investigations ; elles sont prélevées très simplement au ciseau, une trentaine de cheveux doit ainsi être récoltée.
Les tiges pilaires sont évidemment très accessibles aux investigations ; elles sont prélevées très simplement au ciseau, une trentaine de cheveux doit ainsi être récoltée.
Il est parfois nécessaire de faire porter le prélèvement sur des zones variées du cuir chevelu, si les cheveux apparaissent différents suivant les zones considérées.
A – EXAMEN EN MICROSCOPIE OPTIQUE :
Les cheveux sont montés entre deux lames.
Il est parfois nécessaire de les dégraisser, par exemple avec le xylol ou le baume du Pérou.
Aucune fixation n’est nécessaire.
Les deux lames bien solidarisées par l’application d’un ruban adhésif peuvent être conservées indéfiniment.
1- Examen en lumière optique simple :
Il donne déjà certaines indications et fait apparaître les différences de calibre, certaines anomalies de forme, la présence ou l’absence de médullaire.
Le trichogramme, c’est-à-dire le décompte du nombre de cheveux à telle ou telle phase de leur cycle évolutif, apporte quelques renseignements.
Cependant, l’intérêt du trichogramme est limité dans les dysplasies pilaires, au contraire des états alopéciques acquis.
Toutefois, il est utile dans certaines anomalies malformatives où prédomine l’une ou l’autre phase du cycle pilaire.
Il peut être complété par la prise de macrophotographies aux stades de la repousse des cheveux : c’est le phototrichogramme.
2- Lumière polarisée :
La lumière blanche, polychromatique, est constituée de l’ensemble des vibrations vibrant dans tous les plans de l’espace.
L’interposition après la source lumineuse d’un polariseur fait que la lumière est polarisée dans un seul plan, c’est-à-dire que les vibrations se font dans un plan unique.
L’adjonction d’un analyseur permet de voir les rayons lumineux à condition que les plans du polariseur et de l’analyseur ne soient pas perpendiculaires.
En effet, si les plans de polarisation étaient perpendiculaires, le faisceau lumineux serait éteint.
Pour l’examen cristallographique, on utilise justement la position « croisée » du plan de l’analyseur et du polariseur.
L’interposition d’un système polycristallin, tel que le cheveu, entre le polariseur et l’analyseur, va apporter des propriétés optiques supplémentaires.
En effet, le système cristallin qu’est le cheveu, est un milieu anisotrope : il ne restitue pas la lumière polarisée telle qu’il l’a reçue.
Les vibrations lumineuses pénètrent dans le système cristallin du cheveu et y subissent une série de réflexions et de réfractions.
À la sortie de ce système cristallin, les vecteurs lumineux sortent disposés dans deux plans perpendiculaires entre eux qui définissent la biréfringence du cristal : ce sont les « directions privilégiées ».
Si, dans notre outil de travail où polariseur et analyseur sont « croisés », les plans des directions privilégiées se trouvent respectivement dans le plan de l’analyseur et du polariseur, aucun rayon lumineux ne peut émerger : il est nécessaire alors de faire tourner le cristal qu’est le cheveu, sur la platine, pour que celui-ci « s’allume », ses directions privilégiées n’étant pas dans le plan de l’analyseur et du polariseur.
La couleur du rayon lumineux perçu par l’observateur et se propageant dans le plan de l’une ou l’autre des directions privilégiées, est fonction de l’épaisseur et de la nature du cristal traversé.
Telle couleur sera vue en un site précis, si l’épaisseur traversée est un nombre entier de la longueur d’onde vue.
Ainsi, telle ou telle couleur sera perçue par l’observateur en tel ou tel site du cheveu, et le cheveu normal apparaît alors coloré dans son ensemble.
Souvent, cette coloration n’est pas uniforme et un aspect polychrome est noté.
Dans ce cas, cette polychromie est disposée de façon parfaitement symétrique autour de l’axe médian et longitudinal du cheveu.
Pour que l’examen en lumière polarisée rende des services, il faut examiner un grand nombre de segments de cheveu.
Ainsi, les cheveux doivent être coupés en plusieurs segments disposés entre les deux lames de verre.
Les altérations les plus minimes seront donc d’autant plus perçues qu’un plus grand nombre de cheveux ou de segments de cheveux sera examiné.
L’examen en lumière polarisée donne donc les indications à la fois sur l’épaisseur ponctuelle du cheveu et aussi sur sa constitution intime, microfibrillaire.
B – EXAMEN EN MICROSCOPIE ÉLECTRONIQUE À BALAYAGE :
Il apporte la troisième dimension et donne ainsi l’impression de relief.
La microscopie électronique à balayage (MEB) rend compte de la forme de la tige, de l’aspect de la cuticule et de la tranche de section.
La MEB des cheveux ne nécessite ni fixation, ni déshydratation, puisque les cheveux constituent un tissu dur et sec.
La métallisation est nécessaire : elle se fait sous vide et utilise l’or, le vitallium ou l’argent ; la couche couvrante est, suivant les méthodes, de 300 à 500 Å.
Les grossissements peuvent aller jusqu’à 20 000.
Il est nécessaire d’examiner à la fois la tige pilaire sur toute la longueur et également la section.
Le cheveu normal apparaît comme un cylindre à peu près parfait.
De très faibles variations de diamètre peuvent se rencontrer quand on le parcourt sur toute sa longueur.
La couche des cellules cuticulaires enveloppe la tige pilaire.
L’examen en microscopie électronique de transmission est une technique assez délicate du fait de la fragilité de la tige pilaire à la coupe par l’ultramicrotome.
Le cheveu est fixé par le glutéraldéhyde et le tétraoxyde d’osmium, déshydraté par l’alcool éthylique et inclus dans la résine ; il est alors coupé transversalement ou longitudinalement puis coloré par l’uranyle-plomb.
Les coupes transversales doivent être étagées tout le long de la tige pilaire.
L’examen en microscopie électronique de transmission rend compte des différentes couches, médullaire, corticale, cuticule et de leurs composants, trichohyline, kératine, mélanine.
C – ÉTUDE DE LA CUTICULE DU CHEVEU PAR MICROSCOPIE CONFOCALE ET ANALYSE D’IMAGE :
Le microscope laser confocal (Zeiss LSM3) permet d’obtenir des images nettes d’un objet à différentes « altitudes » prédéterminées.
Le cheveu, rendu fluorescent à la rhodamine, est éclairé par une source laser argon 488 nm. Un balayage de l’objet est réalisé tous les 500 nm.
Les séquences d’images obtenues (par exemple 30 séquences) sont récupérées par un logiciel de traitement approprié permettant de procéder à une reconstruction 3D de l’objet examiné.
On obtient à la fois une illustration morphologique de la cuticule du cheveu et la possibilité d’appliquer des paramètres de calcul de rugosité pour caractériser la surface.
Les altérations de la cuticule peuvent ainsi être quantifiées.
D – PESÉE ET COMPTAGE :
La qualité de la repousse de cheveux peut être évaluée par pesée et comptage d’échantillons prélevés tous les 3 mois par exemple, sur une zone exactement repérée.
Le repérage se réalise à l’aide de points de tatouage. Les cheveux sont coupés au ciseau à l’intérieur d’un masque perforé de 1 cm2 appliqué sur le cuir chevelu.
La mèche échantillon est conservée dans une pochette de papier placée dans un pot hermétique.
Chaque échantillon est dégraissé au dichlorométhane, séché puis pesé à l’aide d’une balance de précision 0,01 mg. Une mèche témoin est pesée au même moment et les conditions de température et hygrométrie sont notées : la masse de chaque échantillon sera réajustée en fonction de la masse de l’échantillon témoin.
Les cheveux de chaque échantillon sont comptés par deux expérimentateurs indépendants.
Les échantillons peuvent être conservés pour analyse ultérieure.
E – MESURE DU DIAMÈTRE DU CHEVEU PAR TECHNIQUE VIDÉO :
Le cheveu peut être visualisé par une caméra vidéo munie d’un objectif grossissant.
Un grossissement 100 est un bon compromis entre précision et champ d’exploration.
L’image (noir et blanc) est ensuite numérisée grâce à une carte d’acquisition implantée sur ordinateur (512 ´ 512 pixels, image codée en 256 niveaux de gris).
Le traitement d’image consiste en une recherche des contours par applications successives de filtres permettant de définir les différences de contraste entre le cheveu et le fond de l’image.
Un calcul automatique de distance entre les deux contours est réalisé sur toute la longueur enregistrée du cheveu.
La précision est de l’ordre du micromètre. Le support et l’éclairage doivent être standardisés.
F – MESURE DU DIAMÈTRE DU CHEVEU PAR MICROMÈTRE À LASER :
À l’aide d’un micromètre à laser (Mitutoyut, Japon) le diamètre d’un cheveu peut être mesuré très rapidement et précisément. Un système mécanique fait tourner le cheveu sur son axe longitudinal (Diastront, UK), ce qui permet de calculer des diamètres maximal et minimal et donc la section elliptique.
De telles mesures sont utiles pour :
– l’appréciation de la qualité de cheveux, fins ou épais ;
– l’inclusion dans des tests de propriétés mécaniques ;
– des mesures de gonflement.
Cette dernière méthode consiste à évaluer le taux de gonflement suite à l’immersion du cheveu dans une solution de PH élevé.
Elle permet d’évaluer en particulier l’effet d’oxydation du cheveu (agressions chimiques et agressions solaires).
En effet, les cheveux dont le contenu en cystéine est oxydé en acide cystéique, tendent à gonfler davantage.
G – MESURE DES PROPRIÉTÉS MÉCANIQUES DU CHEVEU :
Quand le cheveu subit une contrainte mécanique, la déformation résultante varie en fonction de son état structurel.
La contrainte la plus utilisée est la traction linéaire.
Le rhéocapillomètre est un appareil qui mesure la force de résistance du cheveu pendant son allongement jusqu’à sa rupture.
En tenant compte de la section du cheveu mesurée au préalable, par exemple par micromètre à laser (Mitutoyut, Japon), la force (N) est exprimée en contrainte (Nm2).
La courbe de rupture obtenue comporte trois zones :
– hookéenne ;
– fluage ;
– après-fluage.
La zone hookéenne représente l’élasticité du cheveu qui dépend notamment des liaisons hydrogène de la kératine alpha.
La zone de fluage reflète la transformation de la kératine entre formes alpha et bêta.
Dans la zone après-fluage, ce sont essentiellement les ponts disulfures qui sont responsables de la résistance à la traction.
H – DOSAGE DES ACIDES AMINÉS DES TIGES PILAIRES :
Pour le dosage de la cystéine et des autres acides aminés dans les cheveux, on procède à une hydrolyse acide pendant 4 heures à 120 °C, suivie d’une chromatographie des acides aminés ainsi libérés.
La relative instabilité de la cystéine dans ces conditions oblige à certaines précautions.
En général, on a recours à une oxydation performique préalable qui transforme la cystéine en acide cystéique, mais plusieurs autres systèmes de dérivation de la cystéine ont été utilisés.
I – IDENTIFICATION DES DIFFÉRENTS TYPES DE KÉRATINE :
On considère que le cheveu contient de 50 à 100 polypeptides différents (kératines ou protéines associées).
Leur séparation est difficile, commençant par une solubilisation laborieuse, puis mettant en oeuvre les techniques analytiques habituelles, essentiellement électrophorétiques.
La biologie moléculaire a partiellement supplanté ces techniques classiques.
En effet, le clonage des gènes codant pour les kératines aussi bien que pour les protéines qui leur sont associées devient de plus en plus un outil incomparable pour déterminer les séquences des différentes chaînes polypeptidiques et, éventuellement, les mutations pathogènes qui peuvent les affecter.
J – DIFFRACTION DES RAYONS X :
Elle permet d’étudier la structure des constituants des cheveux.
En effet, les rayons X, de longueur d’onde donnée et connue, bombardant une structure telle que le cheveu, sont diffusés et diffractés par les électrons qui entourent chaque atome de la structure.
Les constituants à plus haute densité électronique comme les métaux lourds diffractent le plus les rayons X, ceux à plus faible densité diffractent moins.
En recueillant les taches de diffraction à la sortie de la structure examinée, on peut déduire par analyse mathématique l’arrangement des différents constituants.
Au total, cette méthode renseigne sur la constitution des protéines fibreuses du cheveu, et sur la teneur en métaux et métalloïdes.
Le cheveu contient sous forme ionique : calcium, sodium, potassium, zinc, cadmium, manganèse, fer, cuivre, cobalt, magnésium, lithium, plomb, mercure.
Dysplasies pilaires :
A – DYSPLASIES PILAIRES CONGÉNITALES OU ACQUISES, APPAREMMENT ISOLÉES :
1- Monilethrix :
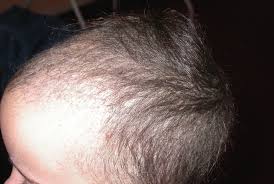
Le monilethrix est une dysplasie pilaire rare de transmission autosomique dominante avec une pénétration élevée et une expressivité variable qui se caractérise par l’aspect moniliforme des cheveux : le cheveu est constitué de nodules elliptiques de 0,7 à 1 mm de diamètre séparés par des espaces internodulaires sans médullaire, ce qui entraîne des fractures du cheveu et par conséquent une alopécie.
Les enfants naissent avec des cheveux normaux.
Le monilethrix apparaît en règle générale vers la 6e ou 8e semaine de vie.
Des cas de révélation tardive à l’âge adulte ont été signalés.
Le diagnostic est évoqué devant l’association de trois signes :
– une alopécie incomplète ;
– des cheveux moniliformes ;
– une kératose pilaire.
L’alopécie est le plus souvent partielle et prédomine sur les zones temporales et occipitales.
Elle peut cependant être diffuse.
Le cuir chevelu est souvent le siège d’une kératose pilaire d’intensité variable.
Cette hyperkératose de l’ostium folliculaire présente un caractère inflammatoire et est due à l’accumulation de cellules cornées.
Les cheveux du monilethrix sont fins, ternes et cassants.
Leur longueur dépasse rarement quelques centimètres (0,5 à 2,5 cm en général).
Le scalp n’est habituellement pas entièrement atteint et l’affection donne volontiers un aspect irrégulier à la chevelure.
Chez certains malades, les cils et les sourcils, les poils axillaires et pubiens, voire même l’ensemble du système pileux, peuvent être intéressés par l’anomalie.
L’examen des cheveux à la loupe montre un aspect caractéristique de chaînette avec des alternances régulières de zones amincies et renflées.
Cependant, le diagnostic de certitude est apporté par l’examen en lumière polarisée qui met en évidence une image spécifique : les variations de calibre de la tige pilaire.
Les zones de resserrement prennent un aspect monochrome blanc ou jaune ; les zones renflées sont régulièrement polychromes.
Cet examen permet de repérer aussi bien les formes majeures de l’affection que des formes cliniquement mineures.
La kératose pilaire peut apparaître avant ou après les signes pilaires et touche le cuir chevelu mais aussi le corps (faces d’extension des membres).
Certains individus d’une même famille peuvent n’exprimer que la kératose pilaire.
Ils peuvent cependant transmettre l’affection à leur descendance.
L’évolution de la maladie est variable.
En règle générale, il existe des améliorations incomplètes à la puberté et chez la femme au cours des grossesses.
Des améliorations transitoires sont signalées par des auteurs pendant l’été.
Le monilethrix est habituellement isolé.
Des anomalies dentaires ou unguéales, une peau hyperélastique, une ichtyose, des syndactylies, une cataracte ou un retard mental ont pu être signalés de manière anecdotique.
Le caractère isolé de la dysplasie pilaire permet de la distinguer des pseudomonilethrix du syndrome de Menkes.
Dans ce cas, les alternances de renflement et de striction sont irrégulières et on retrouve les images plus spécifiques de trichorrhexie noueuse associée.
La pathogénie de la maladie est mal connue.
Les études ultrastructurales ont permis de montrer que les zones renflées ont une structure normale, alors que les zones de striction sont pathologiques.
À ce niveau, il existerait une anomalie de la gaine épithéliale interne, peut-être due à un asynchronisme entre les phases cycliques de prolifération et de repos du follicule pileux.
L’hyperkératose ostiale semble en rapport avec un épaississement de la gaine épithéliale de Huxley.
La normalité du dosage des acides aminés du cheveux exclut a priori une anomalie de leur répartition au niveau des kératines pilaires.
En biologie moléculaire, plusieurs études sont en faveur de différentes mutations d’une kératine de type II, dont le gène est localisé sur le chromosome 12 (12q13).
Cependant, cette anomalie n’est pas confirmée, témoignant peutêtre d’une hétérogénéité génétique de cette maladie.
Le monilethrix est une génodermatose dont le traitement ne peut être que symptomatique et souvent décevant.
En dehors des soins purement cosmétiques, différents traitements ont été proposés et rapportés dans un nombre de cas limité.
Les traitements locaux par émollient kératolytique ou vitamine A restent décevants sur les cheveux, mais peuvent entraîner une amélioration transitoire de la kératose pilaire.
L’étrétinate donne des résultats variables.
L’utilisation de zinc a été proposée par les Toulousains en 1978.
L’efficacité des thérapeutiques est toutefois délicate à apprécier en raison des possibilités d’amélioration spontanée de cette affection.
2- Pseudomonilethrix :
Il a été isolé en 1973 par Bentley-Phillips et Bayles.
L’affection est transmise selon le mode autosomique dominant.
Elle touche exclusivement le cuir chevelu sans kératose pilaire et les nodosites caractéristiques sont disposées irrégulièrement le long de la tige pilaire.
La nature congénitale ou acquise de cette affection a été discutée et certains pensent qu’il ne s’agit que d’un artefact.
3- Hypotrichose héréditaire de Marie Unna :
Il s’agit d’une affection autosomique dominante.
Elle se caractérise par une hypotrichose à la naissance, puis une pousse apparemment normale au cours de l’enfance et enfin une perte progressive des cheveux à partir de la puberté.
Les cheveux sont grossiers et indisciplinés.
Après la puberté, une alopécie s’installe progressivement au niveau du vertex, pouvant évoluer vers une alopécie totale.
Le reste du système pileux est également atteint avec constitution d’une hypotrichose généralisée.
À l’examen microscopique, les cheveux ont un aspect en « fil de fer » ou en « fil électrique torsadé », du fait de la présence de gouttières longitudinales et de torsion selon l’axe, à type de pseudopili torti.
Habituellement isolée, cette dysplasie pilaire a été cependant rapportée, associée à des anomalies unguéales, une kératose folliculaire, un angiome plan, un retard mental ou encore un syndrome d’Ehlers-Danlos.
4- Trichorrhexies noueuses :
C’est la plus fréquente des anomalies de structure de la tige pilaire.
Elle se caractérise par une ou plusieurs petites boules blanchâtres.
Celles-ci sont dues à une fissuration et à une rupture de la cuticule, à travers laquelle les cellules corticales vont faire saillie.
Ces fractures aboutissent souvent à un aspect en « poils de brosse ».
À l’examen microscopique, on constate que les zones dilatées sont dépourvues de cuticules laissant libre accès aux fibres corticales, éclatées en tous sens.
On distingue les trichorrhexies noueuses congénitales et acquises.
On peut également les retrouver de façon sporadique dans d’autres situations, en particulier en association avec d’autres anomalies de la tige comme les pili torti.
* Formes congénitales :
Héréditaires ou non, elles se caractérisent par une distribution généralisée et entraînent une hypotrichose.
Elles peuvent évoquer une acidurie argininosuccinique chez les enfants présentant un retard mental associé à des cheveux courts, irréguliers, râpeux au toucher, friables.
La trichorrhexie noueuse peut se rencontrer également dans le syndrome de Menkes, le syndrome BIDS (brittle hair, intellectual impairment, decreased fertility, short stature) et le syndrome de Tay, qui sont des affections autosomiques récessives, et dans le syndrome de Netherton.
* Formes acquises :
+ Trichorrhexie noueuse proximale :
Elle se rencontre chez des sujets noirs et se manifeste par des plaques d’alopécie au niveau des régions atteintes.
Les cheveux sont si fragiles qu’ils se cassent à quelques centimètres de leur émergence à la suite de manipulations cosmétiques (défrisage, peigne chaud, etc).
+ Trichorrhexie noueuse distale :
C’est une anomalie fréquente qui se rencontre sur des cheveux longs, normaux par ailleurs, chez les Caucasiens et les Asiatiques.
Elle se traduit par de petites taches blanches situées à 10-15 cm de la racine des cheveux.
Les cheveux peuvent être moins brillants, secs, plus clairs à certains endroits, du fait du nombre élevé de petites taches blanches le long des tiges pilaires.
Les dommages cuticulaires peuvent être secondaires aux brossages ou démêlages trop vigoureux, aux expositions solaires prolongées, aux bains de mer répétés, aux traitements capillaires.
5- Pili torti :
C’est une dysplasie rare.
Les tiges pilaires sont rubannées, subissant des torsions régulières de 180° sur leur axe.
La disposition serrée de quatre ou cinq torsions survenant en salves donne un aspect brillant au cheveu et, au microscope, il donne une impression de dilatations moniliformes.
Cette altération augmente la fragilité des cheveux, entraînant donc une hypotrichose et des ruptures localisées ou disséminées en fonction de la sévérité de l’affection et de la zone atteinte.
Les cheveux courts présentent à l’examen un aspect sec et raide alors que la lumière polarisée donne un aspect en paillettes sur le trajet du cheveu.
Plusieurs formes cliniques ont été rapportées.
* Pili torti classique (type Ronchese) :
Cette forme se manifeste, soit comme une dysplasie isolée chez des sujets à cheveux fins et blonds, soit comme un signe d’une dysplasie neuroectodermique, auquel cas elle s’associe à une kératose pilaire, des altérations dentaires, unguéales et cornéennes, une ichtyose, voire même à une surdité neurosensorielle de type cochléaire.
Cette forme est en général à transmission autosomique dominante, mais il existe aussi des cas récessifs et des cas sporadiques.
Il y a une prédominance féminine.
Les cils et les sourcils peuvent aussi être atteints.
Certains cas s’améliorent avec la puberté.
Il n’existe aucun traitement efficace, mais il faut éviter les traumatismes physiques ou chimiques.
* Pili torti associé :
+ Syndrome de Beare :
Autosomique dominant, il se manifeste chez l’adulte à cheveux foncés.
La pilosité faciale et corporelle est faiblement développée, il y a chute des cils et des sourcils, des plaques d’alopécie sur le cuir chevelu, une fragilité unguéale et un retard mental.
+ Syndrome de Björnstad :
Autosomique dominant il associe pili torti et surdité neurosensorielle.
Comme pour le pili torti classique, l’affection s’améliore avec l’âge.
+ Syndrome de Crandall :
Récessif, lié au sexe, il associe pili torti, surdité neurosensorielle et hypogonadisme.
+ Syndrome de Salomon :
Pili torti, dystrophie dentaire et unguéale, nombreuses verrucosités et conjonctivite chronique.
+ Syndrome de Menkes :
Ce syndrome, décrit en 1962 par Menkes, est une affection rare transmise sur le mode récessif lié à l’X.
Cette affection associe pili torti, nanisme, dégénérescence artérielle, hypothermie, lésions osseuses et altérations neurologiques graves : retard mental profond, hypertonie musculaire, convulsions.
Ce syndrome est encore appelé kinky hair syndrome, étant donné que les cheveux caractéristiques du syndrome de Menkes sont bouclés, emmêlés et de couleur blanche.
Par ailleurs, il y a une hypopigmentation cutanée.
Le tableau clinique s’instaure au cours de la première année de vie et aboutit rapidement à la mort.
Les modifications de la chevelure se manifestent vers l’âge de 3 mois lors du remplacement du lanugo.
La symptomatologie neurologique se manifeste entre les 3e et 6e mois de vie.
Ces enfants à peau pâle sont extrêmement susceptibles aux infections qui constituent une cause très fréquente de mortalité.
L’affection est due à une anomalie du métabolisme du cuivre avec blocage d’absorption intestinale du cuivre.
Les taux plasmatiques de cuivre et de céruléoplasmine sont abaissés et il y a déficit de ce métal au niveau du cerveau, du foie, des os, de l’élastine, des poils et cheveux et de la peau.
L’anomalie fondamentale ne résiderait pas seulement dans un défaut de l’absorption intestinale, il y aurait aussi un défaut du transfert intracellulaire du cuivre et de son utilisation, étant donné qu’il existe en excès au niveau de certaines cellules, dont les fibroblastes.
La composition des cheveux en acides aminés est normale mais on a montré une augmentation du nombre des groupes sulfure libre, évoquant une anomalie de la formation des ponts disulfures dans la molécule de kératine.
Le polymorphisme clinique de ce syndrome s’explique par le fait que le cuivre est un cofacteur dans de nombreuses réactions métaboliques.
L’examen microscopique montre des images de pili torti alternantes, caractérisées par des torsions qui alternent régulièrement dans leur orientation.
5- Trichorrhexia invaginata :
La trichorrhexia invaginata ou cheveux en « tige de bambou » correspond à des ruptures de la tige du poil donnant lieu à des renflements dus au télescopage du segment distal dans le segment proximal.
Il existe de nombreux nodules de petite taille siégeant à intervalles réguliers tout le long des tiges pilaires affectées.
La cause pourrait résider dans un défaut passager de la kératinisation de la tige du poil, au niveau où la gaine épithéliale interne est complètement kératinisée, mais où la tige ne l’est pas encore.
La microscopie électronique à transmission montre des images en zig-zag des filaments de l’écorce du cheveu, la portion distale dure de la tige pilaire s’invagine dans la portion proximale qui l’enveloppe.
Les cheveux atteints dans cette affection sont en général courts, fins, cassants, raréfiés surtout sur les zones de frottement.
S’il est possible de rencontrer des cheveux normaux atteints de trichorrhexia invaginata à la suite d’un traumatisme, lorsque la plupart des cheveux présentent des images de trichorrhexia invaginata, il s’agit alors du syndrome de Netherton.
Le syndrome de Netherton est une affection rare de transmission autosomique récessive associant de façon variable, durant son évolution, des manifestations cutanées (ichtyose linéaire circonflexe, lésions eczéma atopique-like, érythrodermie), des anomalies des tiges pilaires (trichorrhexia invaginata le plus souvent) et des manifestations viscérales principalement digestives.
Un taux sérique élevé d’immunoglobulines (Ig)E est fréquemment retrouvé.
Les nodosités des tiges pilaires apparaissent dès l’enfance au niveau du cuir chevelu.
Les cheveux atteints de cette anomalie sont courts, ternes et très fragiles, se cassant sous l’effet de traumatismes (tels le frottement des draps) et n’atteignant jamais une longueur normale.
La portion distale du cheveu peut se détacher et l’extrémité du cheveu prend alors un aspect en « balle de golf ».
Au cours de cette affection, on a rapporté l’association à d’autres dysplasies pilaires : trichorrhexie noueuse, pili torti.
Le traitement du syndrome de Netherton est difficile.
En période néonatale, il faut lutter contre le risque de déshydratation (cutanée ou digestive) et le risque infectieux.
Les traitements locaux (dermocorticoïdes, émollients) améliorent l’état cutané.
On rapporte, dans certains cas, l’efficacité des rétinoïdes systémiques, mais leur utilisation est délicate et ne fait pas l’unanimité.
Plus récemment, des dérivés de la vitamine D locale ont été utilisés et les résultats sont encourageants puisque leur utilisation pendant plusieurs mois semble entraîner une réelle amélioration, sans effets indésirables sérieux.
Il n’existe pas de traitement efficace sur la dysplasie pilaire.
Il faut éviter les traumatismes et veiller attentivement aux soins des cheveux.
6- Syndrome des cheveux anagènes caducs :
Rapporté pour la première fois par Zaun en 1984, le syndrome des cheveux anagènes caducs ou loose anagen syndrome, est une des étiologies d’alopécie non cicatricielle de l’enfant.
Une soixantaine de cas ont été rapportés à ce jour.
Les cheveux s’arrachent facilement et sans douleur et repoussent immédiatement et de façon synchrone.
Il s’observe habituellement chez les sujets blond clair ou blond foncé et affecte les enfants des deux sexes.
Les cheveux sont fins ou de calibre normal, de longueur normale ou diminuée.
On note parfois une mèche de cheveux emmêlés dans la région occipitale.
Les cils et les sourcils sont toujours normaux.
Il n’existe pas d’anomalie associée.
Tosti rapporte cependant un cas d’association avec un syndrome de Nooman.
Le diagnostic est confirmé par le trichogramme, l’examen en lumière polarisée et l’examen au microscope électronique.
Le trichogramme est constitué presque exclusivement de cheveux anagènes dystrophiques, dépourvus de leur gaine épithéliale interne.
L’examen microscopique montre une gouttière longitudinale et une tendance à la triangulation, semblable à ce l’on peut voir dans le syndrome des cheveux incoiffables.
La pathogénie de ce syndrome est une kératinisation anormale de la partie interne des cellules du fond du follicule pileux.
Il pourrait s’agir d’une altération des molécules d’adhésion folliculaires, en particulier la desmogléine, des desmosomes et la cadhérine E.
Il n’existe pas de traitement mais l’aspect des cheveux semble s’améliorer avec le temps.
Le diagnostic différentiel se pose essentiellement avec la pelade.
On conseillera aux parents d’éviter de traumatiser les cheveux par des brushings ou des brossages agressifs.
7- Trichothiodystrophie :
C’est, à ce jour, le marqueur de nombreux syndromes neuroectodermiques.
Les cheveux servent à identifier un groupe de troubles génétiques ayant en commun une altération de la synthèse des protéines riches en acides aminés soufrés (cystine) et un défaut de l’excision/réparation de l’acide désoxyribonucléique (ADN) des fibroblastes après irradiation par ultraviolets (UV).
La trichothiodystrophie (TTD) est une dysplasie pilaire congénitale, transmise selon le mode autosomique récessif, caractérisée par des images pathognomoniques au microscope en lumière polarisée et un déficit en acides aminés soufrés.
Les cheveux sont rares, courts et secs.
Ils sont rugueux au toucher, fragiles et cassent facilement lors de traumatismes même minimes.
La couleur des cheveux est normale.
Les cils et les sourcils sont également atteints.
L’examen microscopique montre des tiges pilaires aplaties rubannées avec des dépressions longitudinales et des phénomènes de tortose.
On note des fractures transversales nettes ou trichoschisis.
En lumière polarisée, les tiges pilaires présentent des alternances de zones sombres et claires disposées en zig-zag et donnent un aspect de tresses ou de « queue de tigre ».
Des images en « habit d’arlequin » et en « queue de léopard » sont également décrites.
L’examen en microscopie électronique à balayage montre une cuticule incomplète ou absente et des sillons longitudinaux.
L’examen en microscopie électronique à transmission montre un arrangement anormal des microfibrilles disposées en « tourbillons » ou en « volutes ».
Le diagnostic est confirmé par l’analyse des acides aminés des cheveux atteints de TTD.
Elle met en évidence une teneur en cystine inférieure de moitié à la valeur normale.
Cette diminution du taux de cystine entraîne une forte réduction de la teneur des protéines matricielles riches en soufre qui sont inférieures à 10 % (normes : 40-45 %).
Il existe une augmentation relative des protéines filamenteuses à faible teneur en soufre.
Les altérations qualitatives ou quantitatives de la composition des cheveux sont très variables en fonction des patients, de minimes à extrêmes, et pourraient traduire des mutations génétiques spécifiques qu’il y aurait lieu de corréler à des phénotypes cliniques.
De nombreux signes cliniques ont été rapportés, associés à la TTD.
Il s’agit surtout d’anomalies touchant le système neuroectodermique.
On a décrit le syndrome BIDS, le syndrome de Tay ou IBIDS syndrome (BIDS syndrome et ichtyose), le PIBID syndrome (IBIDS syndrome et photosensibilité), le xeroderma pigmentosum du groupe D Van Neste a proposé une classification qui regroupe les patients en catégories de gravité croissante :
– A : dysplasie pilaire isolée ;
– B : A + onychodystrophie ;
– C: B + retard mental ;
– D: C + retard de croissance ;
– E : D + ichtyose, type lamellaire congénitale acquise ;
– F:
– E + photosensibilité ;
– déficit de réparation de l’ADN :
– irradiation/UV in vivo ;
– irradiation/UV in vitro ;
– lymphocytes ;
– fibroblastes ;
– complémentation avec xeroderma pigmentosum.
Si la survenue de néoplasies n’est pas associée à la TTD, faisant alors la différence avec le xeroderma pigmentosum, en revanche certains syndromes très complexes sont associés à un très mauvais pronostic de survie dès l’enfance.
8- Syndrome des cheveux laineux ou « wooly hair » :
Il ne s’agit pas exactement d’une dysplasie pilaire entraînant une hypotrichose par rupture des cheveux, mais de cheveux laineux, bouclés et frisés, très fins et clairsemés.
Il en existe trois formes.
* Syndrome des cheveux laineux héréditaire de transmission autosomique dominante :
Il se manifeste dans les premiers mois de la vie.
Cette affection peut s’associer à une trichorrhexie noueuse, un pili torti, un pili annulati et une trichonodose.
Elle intéresse l’ensemble des cheveux.
Dès la naissance ou la première enfance, les cheveux sont frisés, ont une texture fine et sèche qui rappelle la laine de mouton.
Ils sont difficiles à coiffer.
La couleur des cheveux est variable, habituellement sombre.
L’examen au microscope montre une torsion axiale de 180° et parfois une trichorrhexie noueuse.
On a décrit des associations avec une kératose pilaire atrophiante avec ou sans syndrome de Nooman, à des anomalies oculaires (cataracte, rétinite exsudative).
* Syndrome des cheveux laineux familial de transmission autosomique récessive :
Semblable à la forme précédente mais de transmission autosomique récessive.
Les cheveux sont rares, fins, courts, crépus et de couleur claire, parfois très pâles.
L’examen au microscope montre une périodicité de torsion axiale moindre.
Le dommage cuticulaire est important.
* Forme localisée ou « wooly hair naevus » :
Il a été rapporté pour la première fois par Wise en 1927.
Il est d’apparition sporadique. Dans les 2 premières années de vie, on note l’apparition d’une ou de plusieurs zones bien limitées de cheveux crépus.
Ceux-ci sont étroitement enroulés, laineux au toucher et difficiles à coiffer.
Généralement, ils sont plus clairs et plus fins que le restant du cuir chevelu.
Dans de nombreux cas, on a rapporté des associations avec des nævi pigmentaires, épidermiques ou verruqueux linéaires, localisés en général sur les membres supérieurs ou le cou ipsi- ou controlatéral au wooly hair naevus.
9- Syndrome des cheveux incoiffables ou « pili trianguli et canaliculi » :
Ce qui caractérise ce syndrome, c’est la présence de cheveux ébouriffés présents à la naissance ou peu après et qui sont caractérisés par leur caractère « rebelle » à la coiffure.
Les cheveux sont blonds brillants, très clairs.
La structure physique du cheveu, par son caractère triangulaire, ne se laisse pas facilement manipuler.
Les cheveux sont rigides, à l’émergence du cuir chevelu, puis restent dans leur axe dès que la gaine triangulaire formée par la kératine de la gaine épithéliale interne est constituée.
Il n’est pas exclu qu’une anomalie de la papille dermique soit impliquée dans ce processus.
La réflexion de la lumière, particulièrement au niveau des torsions assez fréquentes dans ces cheveux triangulaires, leur donne un aspect brillant argenté.
Cet aspect et les cannelures longitudinales sont bien visibles en microscopie électronique à balayage.
Les premières descriptions ont été faites par Dupré et al (pili canaliculi et trianguli).
Les observations sont généralement sporadiques et certains cas familiaux semblent adopter un caractère de transmission autosomique dominant avec pénétrance variable.
Dans ce cas, on retrouve généralement à l’anamnèse, l’amélioration spontanée de l’affection à la puberté.
Il n’y a pas d’anomalie biochimique caractéristique.
10- Syndrome des cheveux crépus acquis :
C’est une affection rare (acquired progressive kinking of the hair), décrite pour la première fois en 1932 par Wise et Sulzberger.
L’affection débute à la puberté ou quelques années après et surviendrait plus souvent chez les garçons.
Elle est caractérisée par l’apparition progressive de cheveux crépus dans les régions frontopariétales ou du vertex, avec une texture plus grossière, une diminution de la brillance et une difficulté à les coiffer.
Leur couleur peut être plus foncée ou ne pas être modifiée.
De même, la pousse peut être plus lente ou inchangée.
On retrouve parfois un facteur déclenchant comme une radiothérapie, un traitement par rétinoïdes, une permanente ou encore une trichotillomanie.
L’évolution est variable, de l’atteinte progressive de l’ensemble de la chevelure à la régression.
L’examen histologique du cuir chevelu est normal.
L’examen microscopique des tiges pilaires peut montrer des images de pseudopili torti ou des aspects de pili canaliculi.
11- Naevus à cheveux raides :
Le stright-hair naevus est l’apparition d’une zone délimitée de cheveux raides chez un sujet noir dont la chevelure est normalement crépue.
Il peut s’associer à un naevus épidermique.
12- Cheveux noueux ou trichonodose :
C’est une affection acquise, qui est caractérisée par la formation de noeuds.
Elle touche habituellement les cheveux frisés ou bouclés dans leur partie distale et serait due aux frictions.
Elle se voit surtout chez les Noirs.
13- Cheveux fourchus ou trichoptilose :
C’est une anomalie très fréquente qui se rencontre aux extrémités distales des cheveux longs.
Elle correspond aux cheveux fourchus.
Il s’agit d’un clivage, soit à la pointe du cheveu, soit sur sa partie latérale.
Cette anomalie est provoquée par des soins capillaires trop brutaux.
14- Pili bifurcati :
Décrite en 1973 parWeary et al, cette dysplasie se caractérise par la présence de bifurcations intermittentes sur la tige du cheveu qui se rencontrent à des intervalles irréguliers et fusionnent postérieurement.
Chaque branche des bifurcations successives possède sa propre cuticule qui l’entoure complètement.
Il s’agit d’une dysplasie apparemment transitoire, les bifurcations ne se rencontrant que sur un faible pourcentage de cheveux.
Sur le plan clinique, les patients présentent une alopécie diffuse proche de la trichotillomanie ou du pili torti.
La plupart des auteurs pensent que le pili bifurcati ne représente qu’une forme de pili multigemini.
15- Pili multigemini :
Ce terme désigne un groupe de cheveux issus d’un même follicule pileux mais possédant leur propre gaine épithéliale interne.
Ce nom a été proposé en 1951 par Pinkus.
On peut observer deux à huit tiges pilaires qui émergent d’un même canal folliculaire.
Les tiges pilaires sont de diverses formes, liées à la pression s’exerçant entre les tiges pilaires qui partagent le même follicule.
B – DYSPLASIES AVEC ANOMALIES DE COULEUR :
1- Cheveux blancs ou grisonnants :
Ils se rencontrent dans les syndromes de vieillissement prématuré, les albinismes, le piebaldisme, la sclérose tubéreuse de Bourneville, l’ataxie-télangiectasie, parfois dans le syndrome de Rothmund-Thomson, et dans le syndrome de Menkes.
2- Cheveux argentés :
Ils s’observent dans les maladies de Chediak-Higashi et de Griscelli-Prunieras.
3- Cheveux brillants :
* Pili annulati :
Affection sporadique ou familiale (autosomique dominante), elle est caractérisée par une alternance de zones claires et foncées.
Elle se voit surtout sur les cheveux blonds ou peu pigmentés et donne aux cheveux une coloration blond roux avec un aspect moiré. L’affection peut apparaître à la naissance ou au cours de l’enfance.
L’étiologie est inconnue.
À l’examen microscopique, les cheveux présentent une alternance régulière de bandes claires et sombres.
Les bandes correspondent aux zones anormales dans lesquelles la corticale est remplie de multiples petites cavités d’air.
* Pseudopili annulati :
Ils s’observent chez les sujets à cheveux blonds.
Ils n’ont pas de signification.
On note une alternance irrégulière de bandes sombres et claires.
Les bandes sont dues à un effet d’optique en rapport avec un aplatissement du cheveu et une légère torsion (pseudopili torti), qui réfléchit la lumière.
La structure interne de la tige pilaire est entièrement normale.
4- Cheveux « verts » :
Ils affectent les sujets blonds, décolorés ou avec des cheveux blancs.
Ils sont en rapport avec une surcharge en cuivre de l’eau des piscines (souvent inhérente à un excès d’algicides), ou des tuyauteries en cuivre corrodées.
On retrouve fréquemment des facteurs favorisants tels qu’une forte exposition solaire, ou des traitements capillaires oxydants (décolorations, permanentes à froid) qui induisent un accroissement du nombre de sites anioniques forts, avides d’ions cuivre.
Le traitement fait appel aux chélateurs du cuivre (EDTA), utilisés en cosmétologie capillaire comme antioxydants, ou bien aux shampooings à la pénicillamine ou encore à l’utilisation d’eau oxygénée à 3 %.
5- Hétérochromie capillaire :
Rapportée uniquement par les Japonais et dénommées « canities segmentata sideropenica », il s’agit d’une décoloration anormale des cheveux foncés, avec alternance de bandes claires et de bandes de couleur normale le long des tiges pilaires.
L’affection est en rapport avec une anémie ferriprive.
La correction de celle-ci permet le retour à une pigmentation normale.
La pathogénie est inconnue.
C – GÉNODERMATOSES AVEC HYPOTRICHOSE NON CICATRICIELLE :
De nombreuses génodermatoses associent dans leur pathologie une atrichie ou une hypotrichose.
Dans la plupart des cas, il s’agit d’une hypotrichose non cicatricielle, raison pour laquelle nous commençons par l’étude de ces dernières.
Nous ne rapportons ici que les génodermatoses les plus importantes.
1- Altérations squelettiques :
* Hypoplasie des cartilages et des cheveux :
Connue aussi sous le nom de « chondrodysplasie métaphysaire » (syndrome de Mc Kusick), c’est une affection autosomique dominante.
Elle associe nanisme avec déformations squelettiques multiples et une alopécie à cheveux courts, clairs, fins et cassants.
L’alopécie peut être complète dans certains cas.
On rapporte fréquemment d’importantes anomalies du système immunitaire.
L’examen au microscope électronique montre des cheveux normaux mais de petit diamètre.
* Hypotrichose-syndactylie-rétinite :
Polydysplasie complexe à transmission autosomique récessive.
* Syndrome trichorhinophalangien :
L’alopécie constante du cuir chevelu, de la queue des sourcils s’associe à un nez en « poire », une clinobrachydactilie et des malformations osseuses.
Le mode de transmission est variable : autosomique et dominant dans le type I correspondant à une délétion en 8q24.12, ou délétion plus importante dans le type II, 8q24.11-q24.13.
* Syndrome de Pierre Robin :
Alopécie diffuse, division palatine, hypoplasie mandibulaire et glossoptôse.
* Syndrome cardiocutanéfacial :
Il se caractérise par quatre signes :
– anomalie psychomotrice ;
– anomalie cardiaque ;
– faciès typique ;
– anomalies cutanée et pileuse.
Les cheveux peuvent être rares, frisés, de calibre variable avec une ligne d’implantation occipitale basse et des cils et sourcils raréfiés ou absents.
* Syndrome ACD et retard mental :
Autosomique, récessif, il associe atrichie, contractures articulaires et nanisme.
* Syndrome oculodentodigital :
C’est une dysplasie ectodermique de transmission autosomique dominante.
* Syndrome de Dubowitz :
Ce syndrome à transmission autosomique récessive se caractérise par un retard de croissance intra-utérin et postnatal avec nanisme, photosensibilité, microcéphalie et faciès typique : blépharophimosis, micrognathie, associés à une alopécie des sourcils et une hypotrichose à cheveux fins.
* Syndrome de Noonan :
De transmission autosomique dominante, ce syndrome se caractérise par des manifestations cliniques d’un syndrome de Turner mais sur un caryotype normal.
2- Altérations ectodermiques :
Ces dysplasies font l’objet d’un chapitre distinct.
3- Altérations neuroectodermiques :
* Trichothiodystrophie
* GAPO syndrome :
Ce syndrome extrêmement rare associe retard de croissance, alopécie, pseudoanodontie et atrophie optique d’installation progressive.
L’hypothèse actuelle serait un mode de transmission autosomique et récessive.
4- Altérations chromosomiques :
* Syndrome de Down :
Au cours de la trisomie 21, on retrouve souvent une alopécie diffuse avec cheveux fins et hypopigmentés.
* Syndrome de Klinefelter :
Ce syndrome, qui correspond au caryotype 47XXY, est associé à une hypotrichose corporelle.
* Syndrome de Turner :
L’anomalie chromosomique la plus fréquente correspond au caryotype 45X, mais on peut retrouver des mosaïques ou des lésions partielles d’un chromosome X.
5- Altérations du métabolisme des acides aminés :
* Citrullinémie :
Cette affection autosomique récessive est due à un déficit en arginosuccinate- synthétase.
Les cheveux sont courts, brillants, à cuticules interrompues.
* Maladie de Hartnup :
Transmission autosomique récessive.
Anomalie de l’absorption du tryptophane.
* Homocystinurie :
Autosomique et récessive.
Anomalie du métabolisme de la méthionine due à un déficit enzymatique.
* Phénylcétonurie :
* Tyrosinémie :
6- Autres génodermatoses avec hypotrichose :
* Progéries :
Les progéries s’accompagnant d’une alopécie.
* Syndrome de Werner :
De transmission autosomique récessive.
* Syndrome de Hutchinson-Gilford :
Génodermatose à transmission autosomique récessive.
* Pityriasis rubra pilaire :
Dans le type II, soit la forme atypique de l’adulte, l’éruption ichyiosiforme est associée à une alopécie partielle et dans le type IV (forme circonscrite juvénile), la présence de papules folliculaires au niveau des glabelles entraîne une alopécie des sourcils très caractéristique.
* Érythrodermie ichtyosiforme congénitale :
Dans la forme d’érythrodermie ichtyosiforme congénitale sèche de transmission autosomique récessive, il peut exister une alopécie cicatricielle.
* Poïkilodermie congénitale :
La poïkilodermie atrophiante vasculaire congénitale s’associe à une dystrophie unguéale, à une hypotrichose.
Des complications osseuses, ophtalmologiques, un hypogonadisme et un nanisme peuvent survenir.
* Dyskératose congénitale :
Leucomélanodermie, leucokératoses buccales, kératoses diverses, s’associent à une hypotrichose et parfois à un pseudomonilethrix.
Une hémopathie peut survenir proche de l’anémie de Fanconi.
* Épidermolyse bulleuse simple (syndrome de Kallin) :
Cette épidermolyse bulleuse simple (EBS) est associée à des altérations des dents, des ongles et des cheveux.
L’hypotrichose est diffuse, avec des cheveux fins et cassants.
Il existe aussi des plaques d’alopécie non cicatricielle sur le cuir chevelu.
7- Génodermatoses avec hypotrichose et tumeurs :
* Syndrome de Rombo :
Autosomique dominant.
Les sujets présentent une hypotrichose associée à une chute des cils, des papules jaunâtres parfois folliculaires sur le visage et de nombreux trichoépithélium et carcinomes à cellules basales.
* Syndrome de Bazex-Dupré-Christol :
Cette génodermatose rare associe atrophodermie folliculaire, hypotrichose et développement de nombreux carcinomes basocellulaires.
Des travaux récents mettent en évidence une association avec le chromosome X et une mutation dans la région Xq24-q27.
D – GÉNODERMATOSES AVEC ALOPÉCIE CICATRICIELLE :
La liste des affections ci-dessous n’est pas exhaustive.
1- Maladie de Darier :
Affection autosomique dominante caractérisée par une perte d’adhésion entre les cellules épidermiques et une anomalie de la kératinisation.
Le gène de la maladie de Darier a été localisé sur le chromosome 12 et la mutation semble plus précisément se situer en 12q23-24.1.
* Ichtyose liée au sexe :
Cette ichtyose se transmet sur un mode récessif lié à l’X.
* Épidermolyse bulleuse dystrophique :
Les formes bulleuses ou dystrophiques peuvent être à l’origine d’une alopécie lorsque les lésions siègent au niveau du cuir chevelu.
L’anomalie en cause dans ces génodermatoses serait une anomalie du collagène VII.
2- Incontinentia pigmenti :
Dermatose rare liée à l’X.
Au cours de l’évolution de l’affection, les lésions verruqueuses lichénoïdes du cuir chevelu laissent, par involution, une alopécie cicatricielle.
* Chondrodysplasie ponctuée :
Les chondrodysplasies ponctuées (CP) sont des affections associant des manifestations osseuses, oculaires et cutanées.
On distingue quatre types de CP :
– CP de transmission autosomique dominante (syndrome de Conradi-Hünermann) ;
– CP autosomique récessive. Létale dans les premières années de vie ;
– CP récessive liée à l ’X ;
– CP dominante liée à l’X (syndrome de Happle).
Décrite en 1977, caractérisée par des lésions ichtyosiformes, une atrophodermie folliculaire et une alopécie cicatricielle. Les lésions suivent les lignes de Blachko.
On retrouve aussi des anomalies ophtalmologiques et squelettiques ; les calcifications punctiformes des épiphyses sont caractéristiques mais transitoires, disparaissant au cours de l’enfance.
Cette génodermatose est létale pour les embryons mâles hémizygotes.
Le gène responsable n’a pas encore été identifié.
3- Hypoplasie dermique en aires (syndrome de Goltz) :
Ce syndrome associe de multiples anomalies cutanées et mésenchymateuses.
Le mode de transmission serait dominant lié à l’X.
Cette affection est létale chez l’embryon mâle en l’absence de mosaïque, mais une hétérogénéité génétique a été suggérée dans certains cas.







")


")
{kind=link}