



















Introduction :
A – DÉFINITION :
Le chondroblastome est une tumeur osseuse bénigne cartilagineuse constituée de cellules chondroblastiques avec présence de substance chondroïde.
On y rencontre en outre des cellules géantes de type ostéoclastique, des foyers de calcification et dans 10 à 20 % des aspects histologiques de kyste anévrismal.
B – TERMINOLOGIE :
La première description revient à Kolodny en 1927 qui en faisait simplement une tumeur cartilagineuse contenant des cellules géantes.
Ewing, en 1928, parlait de tumeur à cellules géantes calcifiante et Codman, en 1931, de tumeur épiphysaire chondromateuse à cellules géantes.
L’appellation de « chrondroblastome bénin », due à Jaffé et Lichtenstein en 1942, est également retenue par la nomenclature de l’Union de lutte contre le cancer depuis 1965 afin de bien le distinguer de la tumeur à cellules géantes et du chondrosarcome.
Dans la classification internationale de l’Organisation mondiale de la santé (OMS) (1972), cette tumeur est aussi dénommée « chondroblastome bénin épiphysaire ».
C – ÉPIDÉMIOLOGIE. FRÉQUENCE. ÂGE. SEXE :
Le chondroblastome est rare puisqu’il ne représente que 0,5 à 1 % des tumeurs osseuses primitives biopsiées et 9 % des tumeurs osseuses bénignes.
Il a une préférence pour le sexe masculin (2 à 3/1), en général entre l’âge de 10 et 20 ans.
Il s’agit donc d’une tumeur de sujet jeune, exceptionnelle dans la première enfance et après l’âge de 30 ans.
Localisations :
Le chondroblastome siège typiquement sur une épiphyse ou sur une apophyse toujours à proximité d’une physe fertile.
Il peut s’étendre ensuite à la métaphyse en traversant le cartilage de croissance pour devenir épiphysométaphysaire.
Il est exceptionnel que son siège soit purement métaphysaire ou métaphysodiaphysaire.
Il siège dans 80 % des cas à l’extrémité d’un os long avec une prédilection pour l’épaule, le genou et la hanche.
Les localisations les plus fréquentes sont : l’extrémité supérieure de l’humérus (21 %), l’extrémité supérieure du fémur (15 %), l’extrémité inférieure du fémur (16 %), l’extrémité supérieure du tibia (20 %).
La tumeur se situe sur l’épiphyse mais peut aussi se rencontrer sur le trochiter et le grand trochanter.
Le chondroblastome peut se développer plus rarement sur d’autres segments squelettiques : calcanéus, talus, extrémité supérieure du péroné, colonne vertébrale, côtes ou crâne.
Sur le bassin, il peut siéger près du cartilage en « Y » ou sur l’omoplate près de la cavité glénoïde. Une observation avec une atteinte multifocale bénigne a été signalée dans la littérature.
Étude clinique :
Les signes révélateurs du chondroblastome sont souvent discrets et non spécifiques.
Les douleurs sont banales, parfois signalées à l’occasion d’un traumatisme sportif.
Une tuméfaction peut être perçue dans les localisations superficielles comme l’épaule ou le genou.
La boiterie peut être le seul signe d’appel pour une localisation sur un membre inférieur.
La survenue d’une fracture pathologique est une éventualité tout à fait exceptionnelle.
Plus fréquents sont les symptômes qui témoignent d’une irritation articulaire comme une diminution de la mobilité ou l’apparition d’une hydarthrose.
Il arrive aussi que la tumeur soit découverte de façon fortuite à l’occasion d’une radiographie.
L’examen clinique est par ailleurs normal et il n’est retrouvé aucune anomalie biologique.
Étude radiologique :
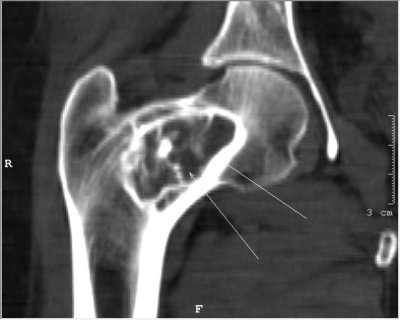
L’étude radiologique standard est souvent suffisante pour évoquer le chondroblastome.
Il réalise une zone d’ostéolyse de forme arrondie ou ovalaire bien limitée par une bordure de sclérose qui témoigne de son évolution lente.
Elle est excentrée par rapport à l’épiphyse, proche d’une corticale ou de l’articulation, et comporte de petites ponctuations calcifiées dans la moitié des cas.
Le cartilage de croissance est souvent visible et peut être traversé par la lésion.
Le chondroblastome est habituellement de petite taille, entre 1 et 6 cm de diamètre, mais il arrive, surtout en cas de récidive, que la tumeur ait une extension supérieure, qu’elle puisse souffler l’os et même envahir l’articulation voisine.
L’image radiologique peut être moins typique lorsque les contours sont mal limités avec des cloisons de refend et une réaction périostée.
La scintigraphie osseuse, si elle est demandée, montrerait une plage très légèrement hyperfixante.
L’imagerie par résonance magnétique (IRM) peut être utile pour apprécier les relations de la tumeur avec le cartilage de croissance, l’articulation et les tissus mous.
Anatomie pathologique :
Même si l’image radiologique, du fait du siège épiphysaire de la lésion, est souvent évocatrice chez l’enfant, la biopsie est indispensable pour affirmer le diagnostic de chondroblastome et ceci encore plus chez l’adulte où le risque de confusion avec un chondrosarcome à cellules claires est important.
Cette biopsie peut constituer également pour des lésions de petite taille, lorsque l’exérèse est totale, le seul traitement.
Dans les autres cas, elle est un préalable obligatoire à tout traitement plus invasif.
A – ASPECT MACROSCOPIQUE :
La tumeur est de consistance ferme, granuleuse, de coloration rosée ou grisâtre.
On y distingue des zones blanchâtres ou crayeuses correspondant à des plages cartilagineuses ou à une imprégnation calcaire et des zones hémorragiques qui évoquent un kyste anévrismal.
La lésion est proche du cartilage de croissance mais elle peut aussi décoller le cartilage articulaire ou même envahir la cavité articulaire.
B – ASPECT MICROSCOPIQUE :
La tumeur est caractérisée par la présence de chondroblastes et de cellules géantes, et par la production de substance chondroïde.
Les cellules chondroblastiques sont polygonales, à bordure nette, munies d’un seul noyau rond ou ovale qui occupe la moitié de la cellule.
Le cytoplasme est granulaire, riche en glycogène et comporte des figures de mitoses normales.
Entre les cellules, la substance fondamentale est pauvre, les fibrilles collagènes sont rares.
Il existe par-ci par-là des foyers de différenciation chondroïde avec des zones de calcification et des zones de nécrose.
Ces dernières sont parfois remaniées par un tissu conjonctif hyalin et un tissu chondroïde.
On trouve au sein de ces zones de remaniement un grand nombre de cellules géantes multinucléées isolées ou regroupées en amas.
Parfois, il existe en périphérie de la lésion des cavités hémorragiques semblables à un kyste anévrismal.
Il existe enfin de possibles associations et des formes de transition entre le fibrome chondromyxoïde et le chondroblastome, du fait de leur parenté histologique.
Sur le plan de l’histogenèse, la cellule chondroblastique peut être rapprochée d’une cellule cartilagineuse adulte et non d’un chondroblaste tumoral.
Elle réalise en effet un aspect de cartilage normal.
Ce fait évoque non pas une transformation de la cellule cartilagineuse mais plutôt une perturbation du processus d’ossification enchondrale.
Diagnostic différentiel :
Les images radiologiques sont souvent évocatrices mais doivent faire discuter d’autres diagnostics que le chondroblastome.
– L’abcès chronique de l’os de siège métaphysaire est entouré d’une épaisse zone de nécrose, il peut contenir des séquestres, mais l’infection peut diffuser au travers du cartilage de croissance et se développer dans l’épiphyse.
– Le kyste anévrismal, le kyste osseux essentiel ne se discutent pas chez l’enfant car il s’agit de lésions métaphysaires ou diaphysaires de plus grande taille.
– Le chondrome sous-périosté réalise une encoche corticale et ne siège pas dans l’épiphyse, même s’il est proche du cartilage de croissance.
–
La synovite villonodulaire peut réaliser, comme d’autres affections synoviales, des images kystiques à la périphérie de l’épiphyse, mais les érosions sont uniquement corticales.
– La tumeur à cellules géantes est exceptionnelle chez l’enfant mais elle doit être évoquée chez l’adulte.
Le contour en est moins net, la taille plus importante et il n’existe pas de calcification intratumorale.
– Le chondrosarcome à cellules claires peut être trompeur chez l’adulte et la biopsie est ici toujours nécessaire.
L’examen histologique peut faire discuter certains aspects frontières avec le kyste anévrismal et le fibrome chondromyxoïde.
La tumeur à cellules géantes est facilement éliminée par les caractères des chondroblastes et l’absence de substance chondroïde.
Le chondrosarcome est reconnu du fait des anomalies nucléaires et des mitoses atypiques, mais des difficultés existent avec le chondrosarcome à cellules claires et le chondrosarcome mésenchymateux qui doivent faire recourir aux tests d’immunomarquage par la protéine S-100.
Le diagnostic de chondroblastome peut donc parfois poser de réelles difficultés, obligeant à une confrontation des données cliniques et paracliniques, voire à recourir à une nouvelle biopsie.
Évolution et pronostic :
L’évolution du chondroblastome est habituellement lente.
Les modifications radiologiques sont souvent minimes à plusieurs mois d’intervalle et il est possible que la lésion ne soit reconnue qu’après de nombreuses années.
À l’inverse, l’évolution peut être beaucoup plus rapide dans certains cas avec un envahissement et une déformation de la totalité de l’épiphyse.
La guérison est obtenue dans la majorité des observations après un traitement chirurgical correct.
Au cours de l’évolution, il existe deux risques : l’un, assez fréquent, est une récidive locale après traitement ; l’autre, rarissime et discutée, est la transformation maligne.
A – RÉCIDIVE :
Le taux de récidive est variable selon les auteurs : 5,7 % pour Schajowicz, 15 % pour Meary et jusqu’à 38 % pour Huvos.
La récidive s’effectue dans l’os adjacent à la tumeur initiale, dans les parties molles et, plus rarement, dans l’articulation.
Ce risque serait plutôt rencontré dans les formes avec une composante anévrismale importante.
En revanche, il ne semble être en relation ni avec l’âge du patient ni avec le siège de la tumeur, ni même avec le traitement pour certains auteurs.
L’atteinte du cartilage de conjugaison a pu, dans certains cas, conduire à un trouble de croissance du segment osseux et à une brièveté du membre.
Il existe des formes agressives pouvant s’étendre à un os de voisinage, par exemple du talus vers le calcanéus ou d’une vertèbre cervicale à la vertèbre sus- ou sous-jacente.
B – RISQUE DE TRANSFORMATION MALIGNE :
Il est très controversé, concerne certains cas avec dégénérescence locale d’une part et la survenue de métastases pulmonaires à distance d’autre part.
Il semble maintenant établi que ces soi-disant chondroblastomes « malins » correspondent en réalité à un chondrosarcome non différencié ou à un chondrosarcome à cellules claires confondu au départ avec un chondroblastome.
Pour d’autres observations, il s’agirait d’une transformation sarcomateuse après une radiothérapie sur une lésion bénigne.
L’apparition de métastases pulmonaires au cours d’un chondroblastome a été signalée dans plusieurs cas.
L’ablation de la lésion pulmonaire, toujours histologiquement bénigne, a conduit à la guérison.
Traitement :
Il s’agit toujours d’un traitement chirurgical de type curetage-comblement.
Il doit comporter un curetage soigneux de la cavité avec l’ablation de la couche osseuse qui correspond à la paroi de la tumeur.
Il est souhaitable, pour certains auteurs, de réaliser en complément un lavage par le phénol ou l’alcool, une cautérisation thermique ou par cryothérapie, ou même le comblement par du ciment acrylique.
Ce dernier doit être évité chez l’enfant.
Il est préférable de réaliser le comblement par une autogreffe corticospongieuse.
Le curetage-comblement par voie arthroscopique a été proposé pour certaines localisations comme le condyle fémoral.
Pour les lésions siégeant sur la tête fémorale, l’abord peut s’effectuer soit par arthrotomie classique, soit au travers du col à l’aide d’une tréphine et de longues curettes sous contrôle scopique.
La résection segmentaire est possible dans certaines localisations (côte, arc vertébral postérieur, péroné).
Elle est surtout envisagée en cas de lésion très étendue ou en cas de récidive, mais il est alors nécessaire de prévoir une reconstruction osseuse ou ostéoarticulaire.
La radiothérapie a pu être proposée pour des lésions d’accès difficile mais elle est à déconseiller.
Elle est formellement contre-indiquée chez l’enfant en raison des risques d’altération des zones de croissance, de la survenue d’un sarcome radio-induit, d’autant que la radiosensibilité du chondroblastome semble incertaine.
Une bonne expérience du traitement des tumeurs osseuses permet aujourd’hui de réaliser une exérèse chirurgicale satisfaisante de ces tumeurs bénignes dans toutes les localisations.
Conclusion :
Le chondroblastome est une tumeur cartilagineuse épiphysaire bénigne qui peut s’étendre vers la métaphyse à travers le cartilage de conjugaison ou plus exceptionnellement vers l’articulation. Elle survient surtout au cours de la deuxième décennie de vie.
Elle siège préférentiellement sur l’extrémité supérieure de l’humérus, du fémur et du tibia.
La radiologie standard est souvent suffisante pour envisager le diagnostic que l’on doit toujours confirmer par biopsie.
Cette biopsie est obligatoire car l’image peut être confondue avec une tumeur à cellules géantes ou avec un chondrosarcome chez l’adulte.
Le traitement est chirurgical.
Il consiste en un curetage complet suivi d’un comblement par des greffons osseux.
Il existe une possibilité de récidive locale bénigne.
La transformation maligne correspond probablement à une tumeur initialement maligne ou à un sarcome radio-induit non reconnu dans certaines formes difficiles.
La survenue de métastases pulmonaires histologiquement bénignes est exceptionnelle au cours du chondroblastome.







")



{kind=link}