



















 Introduction :
Introduction :
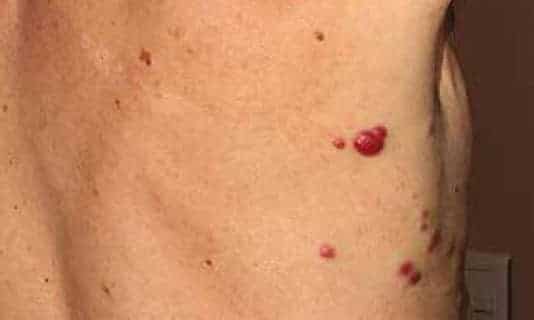
Le carcinome neuroendocrine cutané primitif (CNECP) est une tumeur cutanée rare, initialement décrite par Toker en 1972 sous le nom de « carcinome trabéculaire de la peau », du fait de son aspect histologique.
L’origine cellulaire précise du CNECP est encore débattue, et ceci explique en grande partie les nombreux autres termes utilisés pour le désigner depuis sa description : apudome cutané, tumeur ou carcinome à cellules de Merkel, merkelome, carcinome à petites cellules de la peau, carcinome cutané primitif indifférencié, tumeur neuroépithéliale primitive cutanée et carcinome endocrine de la peau, pour ne citer que les principaux.
La dénomination « carcinome neuroendocrine cutané » apparaît préférable, puisqu’elle est le reflet des caractéristiques ultrastructurales et immuno-histo-chimiques de la tumeur et ne préjuge pas de son origine.
Le diagnostic du CNECP a été facilité par l’apport de la microscopie électronique et surtout de l’immuno-histo-chimie.
Ce cancer était initialement considéré comme faiblement agressif, mais son pronostic s’est secondairement révélé redoutable, avec une propension nette aux récidives locales, locorégionales et aux métastases, ces dernières conduisant le plus souvent au décès du malade.
La rareté du CNECP explique qu’aucune étude prospective comparant l’efficacité des divers protocoles thérapeutiques utilisés aux différents stades de la maladie n’ait encore pu être réalisée, et qu’il n’existe encore aucun consensus sur la conduite thérapeutique à adopter lors de la découverte de la tumeur primitive isolée.
Données épidémiologiques :
L’incidence annuelle du CNECP chez les Blancs, calculée à partir d’un registre regroupant les données d’environ 10 % de la population américaine entre 1986 et 1994, était de 0,35 cas/100 000 chez l’homme et de 0,15 cas/100 000 chez la femme.
De 1991 à 1996, l’incidence annuelle brute, obtenue à partir des données du registre du cancer du Haut-Rhin, variait entre 0,3 et 0,4 cas/100 000 chez l’homme et entre 0,2 et 0,3 cas/100 000 chez la femme.
L’affection atteint principalement des sujets de plus de 65 ans, mais n’épargne aucune tranche d’âge, et des cas ont été décrits chez des enfants dont le plus jeune avait 7 ans.
Les malades sont presque tous blancs et les cas rapportés chez les Noirs, les Asiatiques ou les Polynésiens sont exceptionnels.
Clinique :
Aucun signe clinique n’est spécifique du CNECP.
Les malades consultent le plus souvent en parfait état général, pour un nodule cutané isolé ou une plaque de couleur érythémateuse, violacée ou pourpre, bien limité et mobile par rapport aux plans adjacents.
Quelques cas de tumeur pédiculée ont été rapportés.
La surface est luisante, fréquemment télangiectasique, avec un épiderme parfois ulcéré, mais le plus souvent intact.
La lésion est asymptomatique et mesure en moyenne 2 à 4 cmlors du diagnostic.
Le siège est cervicocéphalique dans au moins 50 % des cas, et alors une fois sur cinq en zone périorbitaire ou palpébrale.
Les autres sites sont, par ordre de fréquence décroissant : les membres inférieurs, les membres supérieurs et le tronc.
Cette dernière localisation représente moins de 10 % des cas. Les muqueuses génitales ou buccopharyngées sont exceptionnellement atteintes.
Quelques cas de localisations cutanées multiples ont été rapportés.
Il peut arriver que le malade consulte pour une adénopathie spécifique, sans qu’une lésion primitive cutanée puisse être découverte.
Les principaux diagnostics différentiels cliniques sont le mélanome achromique, les lymphomes, les métastases cutanées d’autres tumeurs, les tumeurs annexielles, les localisations cutanées de leucémie, les carcinomes basocellulaires et épidermoïdes, les kératoacanthomes, voire les chalazions.
Les anomalies biologiques sont rares, des cas d’élévation des taux sériques d’adrenocorticotrophic hormone (ACTH), de calcitonine, de bêta-human chorionic gonadotrophin (bhCG) ou d’énolase neuronale spécifique (ENS) ont été rapportés
Associations et hypothèses étiologiques :
Au total, 30 à 40 % des malades atteints de CNECP ont eu ou ont, lors du diagnostic, une autre tumeur cutanée considérée comme photo-induite, telle que des kératoses actiniques, une maladie de Bowen, un carcinome épidermoïde, un carcinome basocellulaire ou un mélanome.
Plusieurs cas d’associations, au sein de la même lésion, d’un carcinome neuroendocrine et d’une maladie de Bowen, ou d’un carcinome basocellulaire ou épidermoïde ont même été rapportés.
La survenue préférentielle du CNECP sur les régions photoexposées (85 % des cas), chez des sujets presque exclusivement blancs et ayant fréquemment d’autres tumeurs photo-induites, est en faveur du rôle des irradiations ultraviolettes dans l’apparition de ces tumeurs.
D’autres facteurs doivent cependant exister puisque la lésion peut aussi apparaître sur des zones couvertes, voire des muqueuses.
Quelques cas de CNECP ont été décrits sur des sites précédemment irradiés.
La tumeur s’est parfois développée chez des malades ayant une dysplasie ectodermique congénitale ou une maladie de Cowden.
L’association à des cancers hématopoïétiques ne semble pas exceptionnelle : il s’agit principalement de leucémies lymphoïdes chroniques, de lymphomes, et plus rarement d’autres types de leucémies.
Ces hémopathies pourraient favoriser le développement du CNECP par l’intermédiaire de l’immunodépression qu’elles provoquent.
L’incidence du CNECP paraît d’ailleurs plus importante parmi les transplantés et les malades recevant un traitement immunosuppresseur.
Il semble en revanche que les cas décrits chez les sujets ayant des tumeurs solides telles que des cancers bronchiques, du pancréas, de la prostate ou du côlon, soient plus anecdotiques et liés à la moyenne d’âge élevée de la population atteinte préférentiellement par le CNECP.
Histologie :
Macroscopiquement, le CNECP forme une tumeur non encapsulée, mais bien limitée, ferme et homogène.
L’étude anatomopathologique permet d’observer une lésion située dans le derme, généralement réticulaire, ayant parfois une extension vers l’hypoderme, voire les structures musculaires, ainsi que dans les vaisseaux lymphatiques et sanguins.
Il existe habituellement une zone de séparation avec l’épiderme (Grenz zone) ; quelques cas d’extension intraépidermique, pouvant prendre un aspect pagetoïde, ont cependant été signalés. L’envahissement des annexes pilosébacées est rare.
Les cellules tumorales sont rondes ou ovales, leur taille est relativement uniforme au sein d’une même lésion, avec un diamètre compris entre 12 et 25 µm.
Le cytoplasme est plus ou moins abondant selon le type histologique et le noyau contient une chromatine le plus souvent dispersée en petits amas, mais d’aspect parfois plus dense.
Les nucléoles sont absents ou discrets, typiquement situés à proximité de la membrane nucléaire.
Les mitoses sont habituellement nombreuses, entre 3 et 15 par champ au plus fort grossissement.
Les images de nécrose et d’apoptose sont banales.
Il existe souvent un infiltrat lymphocytaire dense périlésionnel et parfois intratumoral.
Gould et al ont individualisé trois types histologiques de CNECP : trabéculaire, à cellules intermédiaires et à petites cellules.
A – CNECP DE TYPE TRABÉCULAIRE :
Il correspond à la tumeur décrite initialement par Toker en 1972.
Il s’agit en réalité de la forme la moins fréquente, représentant moins de 25 % des cas.
Les cellules s’organisent en travées séparées par un stroma fibreux.
Ces cellules sont rondes ou polygonales, avec un cytoplasme généralement bien défini et abondant.
Elles sont tassées les unes contre les autres, ce qui donne un aspect compact à la lésion. L’activité mitotique est modérée.
Il peut exister, entre les travées, des cavités contenant une substance mucoïde se colorant au bleu alcian. La tumeur est en connexion étroite avec les structures annexielles, en particulier les follicules pileux.
B – CNECP À CELLULES INTERMÉDIAIRES :
C’était le type le plus fréquent dans la série de Gould et al et dans celle de Bayrou et al.
Les cellules se disposent de façon moins compacte que dans le type trabéculaire, leur cytoplasme est aussi moins abondant et moins bien défini.
L’activité mitotique est importante.
L’aspect général peut être assez proche de celui d’un lymphome.
En périphérie de la lésion, on observe régulièrement une organisation trabéculaire.
C – CNECP À PETITES CELLULES :
Dans la forme à petites cellules, il existe un infiltrat diffus, en « nappe », de petites cellules rondes ou fusiformes, au cytoplasme peu abondant, au noyau hyperchromatique, séparées par un stroma abondant.
L’activité mitotique est très intense.
Il existe de larges plages de nécrose. L’aspect est ici voisin de celui des carcinomes bronchiques à petites cellules, ou des carcinomes neuroendocrines d’autre origine.
Ce type peut n’être observé que sur une partie de la lésion et s’associer à un type à cellules intermédiaires.
L’examen histologique standard pose régulièrement des problèmes de diagnostic différentiel avec d’autres tumeurs à petites cellules faiblement différenciées.
La confusion est en effet possible avec une métastase de carcinome bronchique à petites cellules, un lymphome, une leucémie, un mélanome achromique, un carcinome épidermoïde peu différencié, un carcinome annexiel primitif, une histiocytose X, une métastase de tumeur carcinoïde, de carcinome anaplasique, de neuroblastome, de sarcome d’Ewing ou de cancer médullaire de la thyroïde.
Goepfert et al estimaient ainsi que le risque d’erreur était de 66 % lorsque la microscopie optique était utilisée seule pour le diagnostic de CNECP.
Aussi, il faut avoir recours à l’examen en microscopie électronique et/ou surtout à une étude immuno-histo-chimique, pour confirmer le diagnostic.
Microscopie électronique :
Elle a constitué un apport important dans le diagnostic du CNECP, mais le développement des immunomarquages en a réduit l’intérêt.
Cet examen n’est donc plus demandé systématiquement en 2000, puisque la clinique, la microscopie optique, et l’immuno-histochimie suffisent le plus souvent à faire le diagnostic.
Deux éléments ultrastructuraux sont caractéristiques des cellules du CNECP.
Il s’agit d’une part des filaments intermédiaires, et d’autre part des grains neurosécrétoires.
Les filaments intermédiaires sont composés de cytokératines seules ou associées le plus souvent à des neurofilaments et forment des agrégats périnucléaires qui repoussent les autres structures en périphérie du cytoplasme.
Leur diamètre est d’environ 10 nm. Ces filaments, à l’inverse des tonofilaments, ne se raccordent pas avec les systèmes de jonction de la cellule.
Ils ne sont pas détruits par une fixation dans du formol.
Les filaments intermédiaires ne sont par ailleurs observés que dans les métastases de cancers bronchiques à petites cellules et de tumeurs carcinoïdes, avec une fréquence faible dans les deux cas et une disposition différente.
Les grains neurosécrétoires sont des structures arrondies de 80 à 200 nm de diamètre, prédominant en périphérie du cytoplasme.
Ils sont denses aux électrons, avec un halo clair périphérique, et sont limités par une membrane.
Leur nombre est d’autant plus important que la cellule est plus grande et mieux différenciée.
La plus grande partie de ces grains disparaît si la lésion n’a pas été fixée dans le glutaraldéhyde.
Les granules neurosécrétoires des métastases de carcinomes à petites cellules du poumon sont plus petits et dispersés dans le cytoplasme.
Immuno-histo-chimie :
L’étude immuno-histo-chimique constitue une aide importante au diagnostic, notamment par l’expression conjointe et très rare dans d’autres proliférations tumorales de cytokératines et de neurofilaments qui constituent les filaments intermédiaires.
La presque-totalité des CNECP ont un marquage positif pour les cytokératines de bas poids moléculaire 8, 18, 19, et 20, alors que les cytokératines de poids moléculaires plus importants ne sont pas exprimées.
Leur disposition périnucléaire, en motte, est très particulière au CNECP, même si elle peut parfois être observée dans les métastases des carcinomes bronchiques à petites cellules.
La positivité du marquage pour la cytokératine 20 est à la fois sensible et assez spécifique du diagnostic de CNECP.
Cette même disposition périnucléaire est observée pour les neurofilaments, dont le marquage est aussi fréquemment positif dans les cellules tumorales.
Parmi les marqueurs neuroendocriniens, l’ENS est celui qui est le plus régulièrement positif, donnant un marquage diffus de la cellule.
La spécificité de l’ENS est cependant médiocre puisque des métastases telles que celles de cancers à petites cellules du poumon, de tumeurs carcinoïdes et de neuroblastomes ne peuvent être éliminées devant la seule positivité de ce marqueur.
Par ailleurs, la chromogranine A et la synaptophysine, protéïnes présentes dans les grains neurosécrétoires, donnent une réaction positive dans moins de 50 % des cas.
Divers neuropeptides peuvent être exprimés par les tumeurs, quelquefois avec une fréquence faible, et finalement avec une valeur médiocre pour le diagnostic : il s’agit principalement de la calcitonine, du polypeptide intestinal vasoactif, de l’ACTH, de la bombésine, de la somatostatine, de la substance P, de la gastrine, de la metenképhaline et de la leu-enképhaline. L’antigène membranaire épithélial est un marqueur important de différenciation épithéliale.
Il est souvent positif et donne un marquage diffus ou membranaire, mais il est présent dans d’autres tumeurs épithéliales.
L’absence de marquage avec la protéine S 100 est un argument important pour éliminer le diagnostic de mélanome, de même que l’absence de marquage avec l’antigène leucocytaire commun qui élimine le diagnostic de lymphome.
Étude cytogénétique :
Plusieurs anomalies chromosomiques ont été décrites sur les lignées de cellules de CNECP.
Il s’agit le plus souvent d’altérations du chromosome 1 à type de délétion, de translocation ou de trisomie.
Des cas de délétion du bras court du chromosome 3, de trisomie des chromosomes 6 ou 11, et de perte des chromosomes Y ou 13 ou 22, ont aussi été signalés.
Origine du carcinome neuroendocrine cutané primitif :
La plupart des auteurs considèrent que le CNECP dérive des cellules de Merkel.
Ces cellules sont situées, chez l’homme, dans les assises basales et suprabasales de l’épiderme, de l’épithélium des muqueuses, de celui des annexes, et parfois dans le derme où elles forment des complexes avec les fibres nerveuses.
Leur nombre est plus important sur les lèvres, le palais, les paumes et les pulpes digitales.
Le rôle des cellules de Merkel est imprécis.
Leurs rapports étroits avec les axones terminaux des neurones sensoriels cutanés, avec lesquels elles forment parfois de véritables synapses, incitent à penser que ces cellules jouent un rôle dans la perception tactile.
Ce rôle pourrait être celui de mécanorécepteur qui transformerait le stimulus mécanique en signal électrique ou de cellule influençant le seuil d’excitabilité des terminaisons nerveuses.
Il semble aussi que les cellules de Merkel soient impliquées dans le positionnement de ces terminaisons nerveuses. L’origine de la cellule de Merkel fait encore l’objet de controverses.
Néanmoins, l’hypothèse selon laquelle la cellule dérive d’une cellule souche épidermique capable de se différencier en cellules neuroendocrines ou en kératinocytes est actuellement privilégiée par rapport à celles en faisant une cellule dérivée de la crête neurale ou une cellule du système amin precursor uptake and decarboxylation (APUD).
Plusieurs arguments plaident en faveur de l’origine merkélienne du carcinome neuroendocrine.
Il s’agit d’abord de l’aspect très proche des cellules de Merkel normales et des cellules tumorales en microscopie optique et en microscopie électronique, cette dernière révélant des filaments intermédiaires et des grains neurosecrétoires également dans la cellule de Merkel.
Il s’agit ensuite et surtout des caractéristiques immuno-histo-chimiques, avec positivité commune à la fois pour des marqueurs témoignant d’une différenciation épithéliale et pour des marqueurs du système neuroendocrinien.
En effet, la cellule de Merkel exprime les cytokératines 8, 18, 19 et 20, l’antigène de membrane épithéliale, la NSE, les chromogranines et la synaptophysine, de même que certains neuropeptides exprimés par les cellules tumorales telles que la calcitonine, le peptide intestinal vasoactif et la metenképhaline.
Cependant, plusieurs données expliquent qu’il n’y ait pas encore d’unanimité pour considérer que la cellule de Merkel soit à l’origine du CNECP.
D’abord, les CNECP n’ont pas une répartition sur le tégument parallèle à celle des cellules de Merkel, puisqu’ils sont plus fréquents sur la tête, le cou et les membres.
Ensuite, le CNECP est le plus souvent de localisation dermique, avec une séparation nette de l’épiderme, alors que la cellule de Merkel est plutôt intraépithéliale.
Enfin, il existe des différences immunophénotypiques entre les cellules tumorales et les cellules de Merkel.
L’expression des neurofilaments est en effet rare dans ces dernières, à tel point qu’elle fût considérée comme inexistante jusqu’à très récemment ; de plus, la fluorescence obtenue est diffuse et non pas paranucléaire comme dans les cellules du CNECP.
Par ailleurs, des neuropeptides tels que la somatostatine, l’ACTH, ou la substance P n’ont été identifiés que de façon très rare dans les cellules de Merkel humaines normales, alors qu’ils sont plus fréquemment exprimés par les cellules tumorales.
D’autres auteurs considèrent donc que le CNECP se développe à partir de cellules neuroendocrines situées dans le derme, différentes des cellules de Merkel épidermiques, ou à partir d’une cellule souche immature totipotente, qui acquerrait des caractéristiques neuroendocrines lors de sa transformation maligne.
Cette cellule souche pourrait être d’origine épidermique ou annexielle.
Cette dernière théorie pourrait expliquer les cas de tumeurs où coexistent un CNECP et une tumeur épithéliale.
Quelle que soit la nature précise de la cellule à partir de laquelle les CNECP se développent, il semble que sa transformation maligne soit favorisée par l’exposition solaire, les radiations ionisantes et, éventuellement, par des aberrations génétiques qui pourraient altérer le fonctionnement d’un gène suppresseur de tumeur.
Évolution :
Bien que quelques cas de régression spontanée aient été rapportés, y compris au stade de métastases, le CNECP doit être considéré comme une tumeur agressive qui comporte un taux important de récidives locales, locorégionales et de métastases à distance.
Plusieurs études ont montré que la diffusion se faisait le plus souvent par étapes, les malades souffrant d’une maladie disséminée ayant le plus souvent eu, dans un premier temps, une atteinte du premier relais ganglionnaire.
Dans la revue de Shaw et Rumball, 78 des 208 malades (37,5 %) qui avaient une maladie localisée à l’admission étaient décédés au cours d’une durée de suivi moyenne des survivants de 28 mois.
Morisson et al ont trouvé une médiane de survie de 40 mois en l’absence d’atteinte ganglionnaire, tandis que les taux de survie à 5 ans des malades ayant une maladie localisée à l’admission étaient respectivement de 63 et 64 % dans les études de Yiengpruksawan et al et de Eftekhari et al.
Le taux de récidive locale est d’environ 35 % dans l’année suivant l’excision de la lésion primitive.
Dans un cas sur trois, la récidive est multifocale.
Le délai moyen de survenue de ces récidives locales varie de 4,3 à 10,1 mois selon les séries, avec des extrêmes de 1 et 54 mois.
Au total, 75 % des 71 malades qui avaient eu une récidive locale et un suivi de 6 mois au moins dans la série de Hitchcock et al ont développé des métastases ganglionnaires régionales et/ou systémiques. Parmi eux, 55 % sont décédés de leur maladie.
Des métastases ganglionnaires dans le premier relais apparaissent chez la moitié à deux tiers des malades.
Elles sont présentes lors du diagnostic dans 10 à 30 % des cas, sinon elles surviennent en moyenne 7 à 13 mois après le traitement de la tumeur primitive.
Les taux de survie à 5 ans de ces malades étaient respectivement de 48 et 35 % dans les études de Yiengpruksawan et al et de Eftekhari et al, quant à la médiane de survie, elle est de 13 à 34 mois.
Les principaux sites de métastases, au-delà du premier relais ganglionnaire, sont les ganglions, le foie, les os, le cerveau, les poumons, la peau et le tube digestif, mais presque tous les organes peuvent être concernés.
Cette évolution survient chez environ un malade sur trois, dans un délai moyen de 8 à 18 mois et entraîne le décès d’au moins 70 % des sujets dont le suivi est supérieur ou égal à 24 mois.
Le délai moyen entre le diagnostic de maladie disséminée et le décès est proche de 6 mois.
La médiane de survie à ce stade est de 5 à 25 mois.
Sur les 80 malades de Meeuwissen et al, 29 % seulement étaient encore vivants sans avoir eu de rechute 36 mois après le diagnostic.
Hitchcock et al ont trouvé des taux de survie globale de 88 % à 1 an, 72 % à 2 ans et de 55 % à 3 ans.
Certains auteurs ont cependant rapporté des chiffres un peu plus optimistes de 68 % de survie à 3 ans et 64 % de survie à 5 ans, mais sur des échantillons plus faibles, respectivement de 80 et 70 malades.
Environ un malade sur trois décède de sa maladie.
Facteurs pronostiques :
De nombreux auteurs ont tenté d’individualiser l’existence de facteurs pronostiques dans l’évolution des CNECP.
En dehors de l’influence du sexe et du type histologique, les résultats de ces études sont relativement discordants.
Une classification de la lésion dans le groupe des CNECP à petites cellules, la présence d’emboles vasculaires ou lymphatiques ou un index mitotique supérieur à dix mitoses par champ au fort grossissement sont des facteurs histologiques de mauvais pronostic.
Il ressort clairement que le pronostic de la maladie en termes de survie est meilleur chez la femme que chez l’homme.
Hitchcock et al ont trouvé que le taux de survie à 3 ans était de 67,6 % chez les femmes et de 35,6 % chez les hommes.
L’âge ne semble pas influencer l’évolution de la maladie.
Seules, deux analyses univariées, sur de petits effectifs, ont fait ressortir cette variable, mais avec des résultats inverses.
Il en est de même du site initial de la lésion. Si la découverte de métastase(s) systémique(s) est toujours associée à un pronostic mauvais, cela est moins net pour les récidives locales et/ou locorégionales qui, pour Pitale et al notamment, ne semblent pas diminuer la survie.
Une récidive ganglionnaire au premier relais apparaît cependant de mauvais pronostic dans plusieurs séries.
La taille de la lésion lors du diagnostic et sa durée d’évolution avant traitement n’influent sur le pronostic ni pour Pitale et al, ni pour Smith et al, tandis que d’autres études ont trouvé qu’une taille du CNECP supérieure à 2 cm était associée à une évolution plus sombre.
Bilan et suivi :
En dehors d’un examen clinique détaillé et de l’exploitation de tout signe d’appel à la recherche d’une extension locorégionale ou systémique, les examens complémentaires se limitent à une radiographie du thorax.
Ce cliché a surtout pour but d’éliminer la présence d’un carcinome pulmonaire à petites cellules.
Il n’existe pas de consensus sur l’intérêt de réaliser d’autres explorations à titre systémique dans le bilan initial.
Eftekhari et al ont suggéré récemment de réaliser une échographie du premier relais ganglionnaire chez tous les malades et de compléter cet examen par un bilan tomodensitométrique (TDM) ou par résonance magnétique thoraco-abdomino-pelvienne si des adénopathies étaient découvertes.
La scintigraphie à l’octréotide couplé à l’iode 123 ou à l’indium 110, aurait une sensibilité au moins équivalente à la TDM dans le dépistage des métastases du CNECP.
Mais il reste à prouver que la découverte par ces techniques de lésions secondaires encore cliniquement muettes s’accompagne d’une amélioration du pronostic pour le malade.
Il n’y a pas non plus de recommandation officielle pour le suivi des malades considérés en rémission complète, après le traitement d’une lésion primitive.
Certains auteurs préconisent un examen clinique complet tous les mois pendant 6 mois, puis tous les 2 à 3 mois pendant 2 ans, puis tous les 6 mois.
Pour les malades ayant une maladie disséminée, l’évaluation de la réponse au traitement peut se faire suivant les cas par l’examen clinique, la TDM et/ou la scintigraphie à l’octréotide marqué.
Le taux sérique d’ENS pourrait parfois être utile pour évaluer l’évolution de la masse tumorale globale, ou pour surveiller la persistance d’une rémission complète, mais l’intérêt de ce dosage en pratique courante reste à préciser.
Traitement :
L’attitude thérapeutique dépend du stade de la maladie mais aussi, dans bien des situations, de l’expérience de chacun.
En effet, la rareté de la tumeur n’a pas permis jusqu’à présent de conduire d’étude comparant les résultats des différentes modalités thérapeutiques.
A – MALADIE LOCALISÉE AU SITE INITIAL :
Le mauvais pronostic du CNECP, et les difficultés actuelles pour contrôler une maladie ayant dépassé le stade localisé, justifient une conduite agressive au stade initial de l’affection.
Il existe une certaine unanimité pour considérer que le traitement de choix de la lésion primitive est chirurgical et que chaque fois que cela est possible, la marge d’exérèse de tissu sain doit être d’environ 3 cm.
Yiengprunksawan et al ont comparé les taux de récidive locale chez 27 malades ayant eu une marge d’exérèse inférieure ou égale à 3 cm et chez 11 malades avec une marge plus importante. Ces taux étaient respectivement de 15 % et 0 % (p = 0,16).
Cette différence, bien que non significative, a conduit les auteurs à recommander une marge d’exérèse d’au moins 3 cm, attitude qui nous paraît aussi justifiée en l’absence d’essais contrôlés plus larges.
Des auteurs ont proposé d’utiliser la technique de Mohs, en particulier dans les régions où il est difficilement envisageable d’effectuer une exérèse avec la marge souhaitable, comme sur le visage.
Le CNECP est une tumeur radiosensible et, la lésion étant excisée, la plupart des équipes réalisent une radiothérapie sur la cicatrice d’exérèse, ainsi que sa périphérie, afin de réduire le risque de récidive locorégionale et ainsi d’augmenter la survie.
Cette attitude est d’autant moins discutée que la marge d’exérèse est réduite ou que l’examen anatomopathologique révèle l’existence d’une extension lymphatique ou vasculaire.
La dose préconisée est de 50 à 70 Gy à administrer en 20 à 25 fractions, sur 4 à 5 semaines.
La radiothérapie est utilisée seule lorsque l’exérèse chirurgicale n’est pas possible, en raison de la taille et/ou du siège de la tumeur ou du fait de l’état du malade.
Elle peut permettre d’obtenir une rémission complète prolongée.
Les données ne sont pas suffisantes pour proposer de réduire systématiquement les marges d’exérèse et conseiller, par exemple, de n’effectuer qu’une exérèse au ras de la lésion, chez les malades bénéficiant d’une radiothérapie adjuvante.
L’intérêt d’un traitement prophylactique sur les ganglions régionaux est plus discuté.
Plusieurs auteurs préconisent d’irradier, en plus du lit tumoral, le premier relais ganglionnaire et le trajet lymphatique entre la cicatrice d’exérèse et ce premier relais.
La dose préconisée est aussi de 50 à 70 Gy.
Cette attitude méritait d’être évaluée par une étude prospective comme celle menée par M Delaunay (hôpital Pellegrin, Bordeaux), actuellement en cours de réalisation en France.
Le curage ganglionnaire du premier relais n’est le plus souvent réalisé que si des adénopathies sont palpées.
Silva et al recommandaient cependant un curage systématique si la tumeur avait un diamètre supérieur à 2 cm, si l’examen histologique permettait d’observer plus de dix mitoses par champ au plus fort grossissement, ou s’il s’agissait d’un CNECP à petites cellules. Hitchcock et al préconisaient, quant à eux, ce curage systématique si un envahissement vasculaire était constaté, ou si la lésion siégeait sur la tête ou le cou, allant jusqu’à recommander un curage cervical bilatéral radical en cas de siège médian.
Shaw et Rumball jugeaient ce curage souhaitable chez les hommes, les sujets jeunes, et en cas de localisation sur la tête et le cou.
Plus récemment, des auteurs ont suggéré de repérer le ganglion sentinelle, de le biopsier, et de procéder à un curage complet si l’examen histologique révélait son envahissement.
Il reste à prouver que le traitement de métastases infracliniques s’accompagne d’une amélioration du pronostic.
À titre anecdotique, on peut signaler l’obtention d’une rémission complète avec des injections intralésionnelles de tumor necrosis factor a (TNFa) par Ito et al.
Une dose totale de 1,5 million d’unités avait été administrée en 12 jours. Les auteurs disposaient d’un recul de 1 an sans récidive.
B – RÉCIDIVE LOCALE OU LOCORÉGIONALE :
Au stade de récidive locale et/ou dans le premier relais ganglionnaire, les meilleures chances de survie sont obtenues par un traitement associant l’exérèse chirurgicale de la tumeur et le curage ganglionnaire, si les ganglions sont atteints, suivi d’une radiothérapie sur la zone de récidive, et sur l’aire de drainage lymphatique.
Les rechutes après cette association sont moins fréquentes que lorsque la chirurgie ou la radiothérapie sont utilisées seules.
Certains auteurs ont obtenu de bons résultats en administrant dès ce stade, de façon isolée ou en association à la radiothérapie et/ou à la chirurgie, un des protocoles de chimiothérapie utilisé habituellement lorsque la maladie est disséminée.
Il n’est pas possible, devant le faible effectif des malades traités de la sorte, de préciser la place de la chimiothérapie à ce stade de la maladie, et surtout de déterminer le bénéfice supplémentaire qu’elle apporte à l’association radiochirurgicale.
Durand et al ont signalé la régression complète d’une récidive locale traitée par l’interféron a 2b en intralésionnel.
Il n’y a pas eu de récidive avec 1 an de recul.
C – MALADIE DISSÉMINÉE :
Au stade de métastase(s) au-delà du premier relais ganglionnaire, le traitement repose essentiellement sur la chimiothérapie.
La chirurgie et/ou la radiothérapie peuvent lui être associées dans certains cas.
Les drogues les plus utilisées sont celles employées dans le traitement du carcinome pulmonaire à petites cellules. Les protocoles donnant les meilleurs résultats associent : soit doxorubicine (ou adriamycine), cyclophosphamide et vincristine ou prednisone ou dacarbazine ou étoposide (VP 16), soit étoposide et cisplatine, soit vincristine et cisplatine, soit streptozotocine et 5-fluoro-uracile.
Il convient d’associer un traitement préventif du syndrome de lyse tumorale, en particulier chez les malades ayant une maladie très étendue.
Ces traitements permettent régulièrement d’obtenir une réponse partielle ou complète. Dans leur revue de la littérature, Sharma et al trouvaient un taux de 38 % de réponses complètes et de 31 % de réponses partielles avec les protocoles à base de doxorubicine et de cyclophosphamide, avec les protocoles à base de cisplatine, ces taux étaient respectivement de 44 % et 11 %.
Mais ces réponses sont de courte durée dans la majorité des cas, la maladie redevenant évolutive en moyenne en 3,5 à 12 mois.
Les tentatives d’injections intramusculaires d’interféron a 2a ou sous-cutanées d’octréotide n’ont pas donné de résultat satisfaisant à ce stade disséminé de la maladie.
Conclusion :
Le CNECP est une tumeur rare, d’individualisation récente et dont le pronostic est redoutable. Les études dont nous disposons actuellement sont rétrospectives et comprennent des effectifs le plus souvent faibles.
La mise en place de protocoles thérapeutiques prospectifs multicentriques, destinés à définir les meilleurs choix à chaque stade de la maladie, devrait également permettre d’obtenir des données plus fiables sur les taux de récidive et de survie, ainsi que sur la valeur réelle des différents facteurs pronostiques proposés jusqu’à présent.







")



{kind=link}